
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
22q11.2微缺失综合征(22q11.2 deletion syndrome)是以22号染色体长臂上大约3 Mb大小片段缺失为遗传学基础的一类临床症候群,临床表型多达180多种,个体差异性大,以其不同的临床特征而有不同的命名,如腭-心-面综合征(velocardiofacial syndrome, VCFS),圆锥动脉干-异常面容综合征(conotruncal anomaly face syndrome, CAFS),DiGeorge综合征(DiGeorge syndrome,DGS)等。人群中发病率为1∶4 000[1],但由于其临床表型多样,部分患者病情较轻临床特征不典型,以及分子诊断技术发展的滞后,实际发病率可能更高。该病为常染色体显性遗传,93%的先证者为de novo突变,而7%有遗传背景,患病个体22q11.2缺失遗传给子代的概率为50%[2]。现将在本院就诊的1例22q11.2经典微缺失所致DiGeorge综合征病例报告如下。
患者,男性,6岁9个月,系"发现生长迟缓伴低钙血症6年余"入院,家长代诉身高体重较同龄儿童低,伴低钙血症,智力正常,易哭闹,饮食欠佳,脑垂体正常(报告未见),口服鱼肝油1颗/天,碳酸钙1袋/天治疗。1个月前查PTH低于正常。患儿出生时有难产,生后有新生儿黄疸,既往有腭裂手术史,无明显感染病史。查体:身高108 cm,体重21 kg(患者身高Z评分=-2.5,生长迟缓),神清,精神可,无特殊面容,双肺呼吸音清,未闻及干湿啰音,神经系统检查未发现明显异常,无佝偻病征。辅助检查:血磷3.32 mmol/L,血钙1.56 mmol/L;24 h尿磷14.71 mmol/24 h,24 h尿钙0.17 mmol/24 h;25羟维生素D测定29.730 ng/ml;全段PTH<3.00 ng/L;骨代谢标志物测定:N-MID骨钙素84.96 ng/ml,总Ⅰ型胶原氨基酸延长肽994.30 ng/ml,β-胶原特殊序列2 673.00 pg/ml;T细胞亚群:CD3+59.8%,CD3+CD4+33.5%,CD3+CD8+23.9,CD3+CD4+/CD3+CD8+ 1.40。左腕关节正位片显示左腕可见6块腕骨。指骨、掌骨、桡骨骨骺未闭合,形态正常。尺骨远端骨骺未出现。左腕关节正位片提示患儿骨龄为5岁。超声心动图显示其冠状静脉窦扩张(提示永存左上腔静脉)且右位主动脉弓。颈部B超提示甲状腺未见明显占位,双侧甲状旁腺未明显显示。胸部CT提示右位主动脉弓,胸腺组织结构不清且体积偏小。
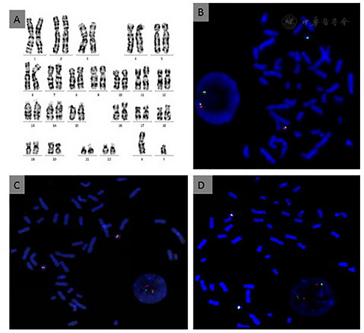
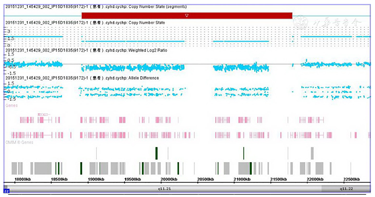
患儿既往有腭裂病史,生长迟缓伴低钙血症,入院后影像学检查提示永存左上腔静脉且右位主动脉弓,双侧甲状旁腺缺如,胸腺发育不良,诊断考虑DiGeorge综合征可能,遂完善该患儿及其父母相关遗传学检测。(1)细胞培养与G显带核型分析,抽取患者外周血进行细胞培养,常规G显带制片、显带与染色收集细胞核型分析。结果如图1A所示,未发现染色体数目或结构异常。(2)荧光原位杂交(fluorescence in situ hybridization, FISH)取患者及其双亲外周血进行细胞培养,制备中期分裂染色体,使用Agilent sure FISH探针试剂盒,红色荧光标记R标记22q11.2(chr22:19,022,989-19,144,127),绿色荧光标记G标记Chr22 CEP(chr22:17, 525,435-18,193,060),计数10个中期分裂象细胞,荧光显微镜下观察荧光信号情况。利用22q11.2和Chr22CEP探针进行检测,结果如图1B、图1C、图1D所示,计数10个中期分裂象细胞,结果均为2G1R,表明患者22q11.2缺失,而患者双亲结果均为2G2R,表明该位点没有发生缺失,患儿该片段缺失为de novo突变。(3)染色体微阵列(Chromosomal microarray analysis, CMA)抽取患者外周血,EDTA抗凝,QIAamp DNA Blood Mini Kit提取外周血样本DNA后采用Affymetrix公司检测试剂盒,使用CytoScanHD/CytoScan750K进行全基因组范围扫描,Affymetrix Chromosome Analysis Suite Software;version 2.0.0.195(r5758)软件分析数据,Affymetrix提供的正常人DNA作为对照标准。经染色体微阵列全基因组扫描,结果如图2所示,证实患儿chr22q11.21区域发生2.9 Mb片段缺失。
患儿DiGeorge综合征诊断明确,入院后予以补钙、维生素D等对症处理。
既往研究发现,22q11.2微缺失综合征一般可分为三种缺失类型,90%的患者缺失区域靠近着丝粒,大约为3 Mb大小的片断,被称为典型缺失区域(TDR),7%的患者缺失片断大小为1.5 Mb,而极少的患者在此区域有更小范围的缺失[3]。染色体22q11.2上有多个低拷贝重复序列(low copy repetitives, LCRs)存在于缺失的3 Mb或1.5 Mb片断的两端,这些低拷贝重复序列有着高度的同源性但非等位,在减数分裂联会同源染色体重组时,可能发生非等位同源重组(nonallelic homologuous recombination, NAHR),造成一条染色体微缺失而另一条微重复[4],或者一条染色体臂内倒位形成环状结构而导致染色体片断缺失。目前在22q11.21-22q11.23区域上共发现8个LCRs,被称为LCR22A-H,可诱导CNVs的各种组合,22DGS/VCFS综合征3 Mb大小的LCR22A-D区间的近端缺失是人类最常见的染色体微缺失[5]。
目前对该综合征病因学研究的重心主要在其缺失区域基因的功能,基因的缺失和剂量不平衡引起临床表型,但缺失区域大小与临床特征之间的具体对应关系仍值得进一步研究[6]。NCBI人类基因组数据提示本研究患儿染色体22q11缺失范围中包括PRODH、HIRA、UFD1L、TBX1、COMT等30个基因。其中TBX1已被证实是22q11.2微缺失综合征最重要的候选基因,在心脏、胸腺、甲状旁腺、腭以及牙齿胚胎发育的基因调节中都起到了不可或缺的作用[7]。心脏流出道、甲状旁腺、胸腺在胚胎发育中都起源于咽弓系统。咽弓主要由内胚层、外胚层、中胚层核心以及咽弓动脉构成,神经嵴细胞迁移分化形成间充质围绕咽弓动脉与中胚层核心。迁徙进入3、4、6咽弓的心脏神经嵴细胞参与主-肺动脉隔形成,协同咽弓动脉的重塑完成心脏流出道的分割[8]。咽弓内胚层以后则发育为甲状腺、胸腺、甲状旁腺及后鳃体[9]。TBX1是T-box基因家族成员,在胚胎发育过程中,该基因家族编码的转录因子在咽中胚层祖细胞中表达,形成一个监管网络以调控心脏和颅面部的正常发育。动物模型表明,TBX1基因的单倍体短缺会导致咽弓及其相关结构的异常生长与重塑[10]。另有研究提示,TBX1基因与miR-96-5p、PITX2共同调节牙齿祖细胞的增殖和腭部发育[11]。本研究患者表型中的右位主动脉弓、胸腺发育不良、甲状旁腺缺如及腭裂均与文献描述相符。60%~75%的22q11.2微缺失综合征患者存在先天性心脏病(CHD)[12],而3%~8%的先天性心脏病患者存在永存左上腔静脉(PLSVC)[13]。永存左上腔静脉与22q11.2微缺失综合征之间的联系已经有文献报道过,但实际上先天性心脏畸形与该综合征之间的关系更为密切[14]。该综合征的患儿中存在明显的神经发育异常的个体占很大一部分比例,并且其中约25%的患儿在青春期或成年后表现为精神分裂症[15]。本研究患者缺失区域中包含的COMT、PRODH、GNB1L、UFD1L、MED15、ZDHHC8、RTN4R、ARVCF等基因都与该综合征精神分裂症的表型相关[16]。由于随访时间较短,尚未发现该患儿有明显的精神异常。
22q11.2微缺失综合征常见的临床表现非常广泛,主要包括:面部畸形、先天性心脏异常、腭咽闭合不全伴或不伴腭裂、胸腺发育不全、免疫缺陷、甲状旁腺发育不全、生长发育迟缓、学习能力低下、精神障碍、肾,眼和骨骼畸形、听力损失、喉畸形等[17]。以胸腺发育不良、低钙血症、先天性心脏病和面部畸形为主要表现的DiGeorge异常根据胸腺受累的严重程度,又分为完全性DiGeorge异常和部分性DiGeorge异常。本研究中的患者虽经胸部CT检查提示胸腺组织结构不清,体积偏小,但患者T细胞亚群仍在正常范围内,病情较轻,无明显感染病史,属于部分性DiGeorge异常。
几种遗传学诊断方法相比较,传统的G显带核型分析虽操作简单、费用低廉,但只能检测出较大片段的染色体缺失或易位以及额外的双着丝粒染色体,对于<5 Mb的缺失常无法检出。FISH分析具有高敏感性高特异性的优点,但操作相对复杂、工作量大且一个探针只能检测一个位点。而染色体微阵列(CMA)具有更高的检出率(25%~30%,而核型分析检出率仅3%~5%),国际上公认作为遗传病检测的一线技术。对于已明确诊断22q11.2微缺失综合征的双亲,可用FISH检测了解其是否也存在染色体微缺失,便于为其再次生育提供遗传咨询。本研究中的患者经CMA检测证实其染色体有2.9Mb大小片段的缺失,而FISH提示患者双亲该位点正常,该片段缺失为de novo突变。
DGS患者的治疗方面,低钙血症可用钙剂和维生素D治疗,T细胞功能缺陷可给予胸腺肽治疗,心脏畸形强调早期诊断和手术治疗,而学习和语言能力的低下则主要依靠早期干预。由于22q11.2微缺失综合征临床表型复杂,国内对本病认识不足,且染色体微阵列等分子遗传学检测价格昂贵、未能建立完善的筛查体系,临床上很多病例不能得到确诊。本病例报道分析了22q.11.2微缺失综合征的临床表现及遗传学诊断,有利于该病患者早期诊断、干预及遗传咨询。
本例患者经积极治疗后病情平稳,一般情况良好,出院后继续口服活性维生素D类似物治疗,我科门诊长期随访。

























