
提高儿科医师对严重联合免疫缺陷病(severe combined immunodeficiency,SCID)的认识,从而加强对SCID的早期甄别及治疗。
回顾性分析我院2007至2018年期间收住院治疗的10例SCID患儿的临床表现、相关免疫学检查结果、影像学表现及转归情况。
SCID的临床表现为出生后短时间内即发生频繁且严重的感染,10例患儿,男8例,女2例,平均发病年龄4.2个月,平均诊断年龄6个月;肺部影像学示8例胸腺影缺如,9例肺CT示重症肺炎,3例并发肺部真菌感染。7例死亡患儿中,6例因感染死亡,死亡年龄均<1岁。实验室检查提示10例患儿均有细胞和体液免疫功能异常;10例CD3+T细胞均<20%,CD16+CD56+(NK%)3例>85%,7例≤2%;7例B细胞反应性增高,但5例分泌免疫球蛋白功能均明显降低。IgG<2.0g/L 7例,>2.0 g/L 3例。8例患儿行基因检测证实为SCID。
SCID的临床表现为生后短时间内即发生频繁的细菌、病毒及真菌感染,在临床工作中对于小婴儿生后反复发生感染,或多部位严重感染且迁延不愈时,要想到SCID的可能并及时行相关免疫功能检查,争取做到早期诊断,及时治疗,尽早行骨髓干细胞移植。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
严重联合免疫缺陷病(severe combined immunodeficiency,SCID)是原发性免疫缺陷病(primary immunodeficiency disease,PID)中的一种,以T淋巴细胞伴或不伴有B淋巴细胞缺乏或功能异常为特征。其发病率非常低,国外文献报道其发病率约为1/58 000[1],我国尚无相关统计数据。该病通常发病年龄较早且临床表现较重,感染多为致死性,若不予以免疫重建治疗,患儿大多于早期死亡[2],因此加强对SCID的早期识别和及时救治非常必要。本研究对中国医科大学附属盛京医院2007至2018年间收住院治疗的10例SCID患儿的临床资料进行回顾性分析,旨在总结各类SCID的临床特点,以利于加强儿科医师对该类疾病的认识,便于早识别和及时救治SCID患者。
选择2007至2018年中国医科大学附属盛京医院住院诊断为SCID的10例患儿为研究对象。本研究经过医院伦理委员会批准及家属知情同意。
(1)明确诊断标准:2岁以内患儿具有经胎盘传递而来的母体T细胞或CD3+T细胞低于20%,绝对淋巴细胞计数<3×109/L,并符合以下至少1项:①细胞因子共有的γ链(γc)基因突变;②JAK3基因突变;③RAG1或RAG2基因突变;④IL2Rα基因突变;⑤ADA活性低于对照的2 %或其2个等位基因均突变。(2)可以诊断标准:2岁以内患儿CD3+T细胞低于20%,绝对淋巴细胞计数低于3×109/L,丝裂原增殖反应低于对照的10%或循环中出现母体淋巴细胞。
血清免疫球蛋白测定采用IgM IMMAGE试剂盒,双光径免疫浊度分析仪(贝克曼公司,美国)进行分析。T、B细胞分化簇(CD)测定采用荧光标记的抗CD19、抗CD3、CD4、CD8、CD56的单克隆抗体(贝克曼公司,美国),采用流式细胞仪进行分析。
提取患儿血DNA标本送至中国香港儿童医院、上海交通大学附属新华医院、上海复旦大学附属儿科医院及北京康旭医学检验所进行基因检测。
采用SPSS18.0统计分析软件,计量资料以均数±标准差( ±s)表示,计数资料以例(%)表示。
±s)表示,计数资料以例(%)表示。
10例SCID患儿中,男8例,女2例,男∶女为4∶1;平均发病年龄4.2个月,平均诊断年龄6个月;10例患儿均诊断肺炎,其中9例患儿肺CT提示重症肺炎(图1),且8例胸腺影缺如,3例(例1、4、9)并发肺部真菌感染;其中1例(例10)注射卡介苗后发热,注射部位红肿破溃,同侧颈部及腋下淋巴结肿大,考虑其为播散性卡介苗病可能(图2);10例患儿中有4例(例1、2、5、10)存在家族史,例1患儿母亲舅父生后不久死亡;例2患儿哥哥于出生3个月时因持续低热、肝脾大、周身皮疹,于当地医院救治无效死亡;例5同胞兄长因重症肺炎于1岁死亡;例10患儿母亲前2个孩子分别于2岁及6个月夭折。本组10例患儿中,7例死亡,1例失访,2例患儿存活,病情好转,已经接受配型,目前等待移植中;死亡的7例患儿中,除例2为造血干细胞移植术后发生免疫排斥反应死亡外,其余6例均为感染后死亡;7例患儿死亡年龄均不超过1岁,见表1。
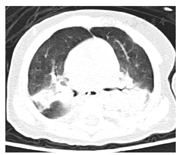
双肺多叶段炎症,局部实变;未见胸腺影。

10例严重联合免疫缺陷病患儿的临床特点
10例严重联合免疫缺陷病患儿的临床特点
| 例号 | 性别 | 发病年龄 | 诊断年龄 | 诊断 | 疫苗反应 | 家族史 | 转归 |
|---|---|---|---|---|---|---|---|
| 1 | 男 | 3.5个月 | 4个月 | 铜绿假单胞菌性脓毒症;急性重症支气管肺炎(细菌混合真菌感染);Ⅰ型呼吸衰竭;面神经核下瘫;鼻瘘;腮裂瘘;多发皮下疖肿;低蛋白血症;混合性贫血(中度) | - | + | 死亡 |
| 2 | 男 | 5个月 | 5.5个月 | 急性重症支气管肺炎 | - | + | 死亡 |
| 3 | 男 | 3个月 | 4个月 | 重症迁延性肺炎;脓毒症(凝固酶阴性葡萄球菌);顽固性口腔溃疡、鹅口疮;心力衰竭;呼吸衰竭 | - | - | 死亡 |
| 4 | 男 | 0.5个月 | 1.5个月 | 重症肺炎(巨细胞病毒感染、真菌感染);肝功能受累 | - | - | 死亡 |
| 5 | 男 | 3个月 | 11个月 | 急性重症支气管肺炎;营养不良 | - | + | 死亡 |
| 6 | 男 | 5个月 | 10个月 | 急性重症支气管肺炎,泌尿系感染 | - | - | 好转,等待移植中 |
| 7 | 女 | 9个月 | 11个月 | 急性重症支气管肺炎;迁延性腹泻病 | - | - | 失访 |
| 8 | 男 | 4.5个月 | 6个月 | 急性重症支气管肺炎;心力衰竭;呼吸衰竭 | - | - | 死亡 |
| 9 | 男 | 6.5个月 | 8个月 | 急性重症支气管肺炎(细菌混合真菌感染);Ⅱ型呼吸衰竭;营养不良 | - | - | 死亡 |
| 10 | 女 | 2个月 | 4个月 | 急性支气管肺炎;播散性卡介菌病(?) | + | + | 好转,等待移植中 |
注:+为阳性。
免疫学检查提示10例SCID患儿均有细胞和体液免疫功能异常;10例CD3+T细胞均<20%,CD16+CD56+(NK%)3例>85%,7例≤2%;7例B细胞反应性增高,但5例分泌免疫球蛋白功能均明显降低。7例IgG<2.0 g/L,3例>2.0 g/L。
基因检测结果:联合中国香港儿童医院、上海交通大学附属新华医院、上海复旦大学附属儿科医院及北京康旭医学检验所对8例患儿进行基因检测(其余2例患儿家长拒绝行基因检测),均证实其为SCID,其中IL2RG基因突变4例,RAG1基因突变3例,JAK3基因突变1例。10例SCID患儿的免疫表型和基因检测结果见表2。
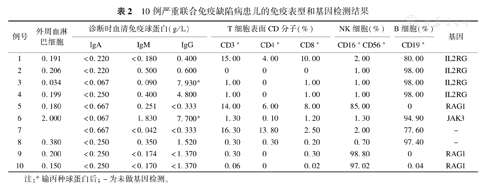
10例严重联合免疫缺陷病患儿的免疫表型和基因检测结果
10例严重联合免疫缺陷病患儿的免疫表型和基因检测结果
| 例号 | 外周血淋巴细胞 | 诊断时血清免疫球蛋白(g/L) | T细胞表面CD分子(%) | NK细胞(%) | B细胞(%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| IgA | IgM | IgG | CD3+ | CD4+ | CD8+ | CD16+CD56+ | CD19+ | 基因 | ||
| 1 | 0.191 | <0.220 | <0.180 | 0.400 | 15.00 | 4.00 | 10.00 | 2.00 | 80.00 | IL2RG |
| 2 | 0.206 | <0.220 | 0.500 | 0.600 | 0 | 0 | 0 | 1.00 | 98.00 | IL2RG |
| 3 | 0.034 | <0.067 | 0.090 | 7.930a | 1.00 | 0 | 1.00 | 1.00 | 98.00 | IL2RG |
| 4 | 0.199 | <0.250 | 0.400 | 4.800 | 1.00 | 0 | 1.00 | 1.00 | 98.00 | IL2RG |
| 5 | 0.180 | <0.667 | 0.251 | <0.333 | 14.00 | 6.00 | 8.00 | 85.00 | 0 | RAG1 |
| 6 | 2.000 | <0.067 | 1.830 | 7.700a | 1.30 | 0.10 | 1.20 | 1.30 | 94.90 | JAK3 |
| 7 | <0.667 | <0.042 | <0.333 | 16.30 | 13.80 | 2.50 | 2.00 | 77.60 | - | |
| 8 | 0.380 | <0.250 | 0.350 | 1.520 | 0.30 | 0.30 | 0.20 | 0.70 | 97.40 | - |
| 9 | 0.200 | <0.250 | <0.174 | <1.370 | 0.30 | 0 | 0.30 | 98.80 | 0 | RAG1 |
| 10 | 0.150 | <0.250 | <0.170 | <1.370 | 0.06 | 0 | 0.02 | 97.02 | 0.04 | RAG1 |
注:a输丙种球蛋白后;-为未做基因检测。
近年来,随着免疫学技术和基因检测技术的迅猛发展,人们对先天性免疫缺陷病的认知不断提升。SCID属于PID中的联合免疫缺陷,此病预后差,多数患儿在1~2岁内死于严重感染,所以SCID是儿科医生必须面对的急症[4]。SCID患儿多年龄小、病情重,本组10例患儿发病年龄最小0.5个月,最大9个月,平均发病年龄仅4.2个月;平均诊断年龄6.5个月;诊断主要以肺炎为主,10例中9例均为重症肺炎,2例存在营养不良,还有1例考虑播散性卡介苗病可能,以上这些数据均与文献报道一致[5,6]。10例SCID患儿中,男8例,女2例,男女比例为4︰1;8例男性患儿中4例(例1、2、3、4)为X-SCID患儿,该4例X-SCID患儿中有明确家族史的仅有2例(例1、2)有阳性家族史,所以在看诊过程中询问患儿家族史很重要,但是没有相关家族史不能排除X-SCID可能。SCID患儿生后不久即发生频繁的细菌、病毒及真菌的致死性严重感染,其他临床表现包括生长发育障碍,持续低毒力的条件致病菌如卡氏肺孢子菌、巨细胞病毒(cytomegalovirus,CMV)及隐孢子虫等感染。本组患儿感染疾病以呼吸道感染为主,10例患儿均患肺炎,其中9例为重症肺炎(图1),亦有患儿存在消化道及泌尿系感染;病原体方面,3例患儿存在真菌感染,还有患儿有细菌(铜绿假单胞菌、凝固酶阴性葡萄球菌)感染,明确CMV感染1例,1例患儿(例10)考虑为播散性卡介苗病(其卡疤部位见图2)。本组患儿病原检出种类与文献相似,但检出率相对较低,可能与检出方法局限且敏感性不高有关。临床医生应根据临床表现给予SCID患儿积极完善相关部位的病原学检测,极力寻找感染的相关证据,继而根据病原学检测结果进行合理抗感染治疗,患儿才能得到合理化的抗生素治疗方案,进而避免抗生素耐药的发生。深部真菌感染对于儿童来说是非常严重的,甚至可以是致死性的,免疫缺陷是高危因素之一[7]。本组10例患儿中有3例(例1、4、9)存在肺部真菌感染,虽应用抗真菌治疗,但仍因感染死亡。我国人群CMV感染率较高,有文献显示1~3岁组儿童CMV感染率高达83.2%[8]。SCID患儿CMV感染可引起严重的脏器损伤,甚至可危及患儿生命。但在SCID患儿中,CMV-IgM可出现假阴性,所以对CMV感染的诊断不能仅依靠CMV-IgM一项指标,还应考虑CMV-DNA的拷贝数和临床表现综合判断,及时进行针对性治疗。接种卡介苗后出现明显不良反应,应考虑患儿为PID的可能,尤其是SCID、慢性肉芽肿病及高IgM综合征。对于SCID患儿,接种卡介苗后最常见的并发症是结核的播散性感染[9],危及患儿生命,并成为以后免疫重建的主要障碍。因此,对家族中有早年夭折史或接种疫苗后有不良反应的患儿应延迟或避免接种卡介苗。
本组前4例患儿均为T-B+NK-表型,基因均证实其为IL2RG突变。此4例患儿淋巴细胞绝对值计数均少于3×109/L,提示淋巴细胞绝对计数是非常有用的SCID筛查诊断方法,与文献[10]相符。本组3例患儿(例3、4、6)免疫球蛋白IgG均>4 g/L,例3和例6为输注丙种球蛋白后所测,例4诊断年龄仅为1.5个月,其IgG偏高考虑与母体子宫内IgG输注残留有关。因此,血IgG无明显降低不能除外SCID。目前,已知的X-SCID致病基因只有IL2RG,已报道的IL2RG突变点达200余种。本组10例患儿中4例为X-SCID,占总数的40%,与文献报道的X-SCID在SCID患儿中的比例相似[11]。本组4例X-SCID患儿符合典型的SCID临床表现,此类患儿若不及时就诊大多于1岁内死亡[12]。重组活化基因RAG1、RAG2编码的蛋白参与T和B细胞抗原受体基因的体细胞重组。该蛋白识别重组信号序列,产生DNA双链断裂,允许V(D)J片段重组,其突变导致抗原受体产生缺陷[13],从而导致常染色体隐性遗传SCID,约占所有SCID的20%[14]。如RAG1、RAG2完全失去活性,将导致典型的SCID临床表型。如果RAG1、RAG2残留部分酶的活性,将导致Omenn综合征[15],其典型的临床表现包括生后不久即发生严重的感染、肝脾和淋巴结肿大、红皮病等。本组3例RAG1基因突变的患儿(例5、9、10)临床表现均为典型的SCID,无Omenn综合征表现。此3例患儿的淋巴细胞绝对值计数和IL2RG基因突变的4例患儿(例1、2、3、4)一样,均少于3×109/L,与文献报道相符[16]。JAK3是细胞质内的一种酪氨酸激酶,与γc物理和功能上相关联,允许细胞因子依赖的信号传导。故此类基因突变患儿的临床表现可与IL2RG缺陷的患儿症状相似。此类缺陷通常会导致常染色体隐性遗传SCID。若患儿体内残留有JAK3蛋白表达和功能,其体循环中可出现自主、活化、寡克隆和功能低下的T淋巴细胞[17]。SCID患儿的基因表型往往决定了其淋巴细胞亚群分布的特点,IL2RG和JAK3所致者多表现为T-B+NK-,RAGl所致者表现为T-B-NK+[18,19],本组中已行基因诊断的8例患儿的淋巴细胞亚群分布特点和基因表现完全符合此特点。
目前治疗SCID最有效的根治手段是异基因造血干细胞移植(hematopoieticstem cell transplantation)。据报道,在北美采用HLA全相合的同胞供者造血干细胞移植治疗SCID的总体存活率达90%以上[20]。有研究显示<3.5个月和未发生感染的SCID患者移植存活率更高。本组10例SCID患儿仅1例(例2)进行了造血干细胞移植,但不幸的是术后发生免疫排斥反应而死亡。绝大多数患儿因诊断延迟,感染严重,脏器功能衰竭和家庭经济原因而错失异基因造血干细胞移植的机会。所以临床医生遇到婴儿早期出现致死性严重感染(尤其是条件性致病菌引起的)或注射卡介苗出现不良反应者,需及时实验室检测免疫功能异常(免疫球蛋白降低、淋巴细胞尤其T细胞缺如及胸腺缺失者),要考虑SCID的可能,尽早完善基因检测确诊,以争取造血干细胞移植的机会。

























