
探讨低氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)在心肌缺血后适应中对心肌的保护作用及机制。
健康成年SD大鼠40只,随机分为4组,每组10只。对照组(A组):仅在冠状动脉前降支穿线,不结扎,持续225 min;缺血再灌注组(B组):阻断前降支45 min后再灌注3 h;缺血后适应组(C组):阻断前降支45 min后,再灌注开始瞬间实施再灌注10 s-缺血10 s,共3个循环的干预,再灌注3 h;缺血后适应+HIF-1α抑制剂组(D组):阻断前降支45 min后,腹腔注射HIF-1α抑制剂AG490(3 μg/g),再灌注瞬间实施再灌注10 s-缺血10 s,共3个循环的干预,再灌注3 h。在结扎冠状动脉前降支前、缺血45 min后、再灌注3 h后3个时间点,在大鼠股静脉处分别测定血清中的肌酸激酶、肌钙蛋白含量。再灌注3 h后,取大鼠心脏用2,3,5-氯化三苯基四氮唑染色法测定心肌梗死面积;Western blot法测定心肌组织中HIF-1α在各组中的表达情况。
(1) 4组大鼠血清肌酸激酶、肌钙蛋白含量在结扎前(术前)差异无统计学意义(P>0.05);缺血45 min后,B组、C组及D组与A组比较差异有统计学意义(P<0.01);再灌注3 h后,B组、C组及D组与A组比较差异均有统计学意义(P均<0.01),且B组、D组显著高于C组(P<0.05)。(2)B组[(45.81±5.96)%]、C组[(37.17±4.99)%]及D组[(45.00±3.29)%]与A组[(2.46±1.13)%]心肌梗死面积比较,差异均有统计学意义(P<0.01);B组、D组与C组心肌梗死面积比较,差异有统计学意义(P<0.05)。(3)心肌组织中,A组HIF-1α蛋白微量表达;B组HIF-1α蛋白表达高于A组(P<0.05);C组HIF-1α蛋白显著高于B组(P<0.05);D组HIF-1α蛋白几乎不表达。
缺血后适应增加HIF-1α在心肌中的表达;HIF-1α表达增加可能参与大鼠心肌缺血后适应心肌保护的过程。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
心肌保护是心脏外科领域的研究热点,冠状动脉搭桥、瓣膜置换以及先天性心脏病手术均涉及心肌缺血再灌注损伤的过程。1977年,Murry等在心肌缺血前施加多次短暂的缺血干预,发现此干预可以引起缺血再灌注后的心肌保护作用[1]。此后,Zhao等[2]通过阻断冠状动脉前降支1 h后,给予与缺血预适应相同的干预,再开放冠状动脉灌注180 min,发现此干预同样具有心肌保护作用,而且发现心肌梗死面积与对照组相比差异有统计学意义,因缺血导致的心肌组织水肿程度明显减轻,因此,他们提出缺血后适应的概念。这个观点的提出解决了缺血预适应存在的因心肌梗死发生的不可预知性的缺点,更利于在临床实践上推广和运用。临床研究中也证实介入治疗增加了缺血后适应的干预效果,且很好地改善了急性心肌梗死患者的预后[3]。近年来越来越多的学者对心肌保护的分子机制进行探索,其中低氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)是众多备受关注的研究对象之一。HIF-1α在低氧条件下能够诱导下游100多个基因的表达,通过不同的机制实现心肌保护作用[4]。但尚未有研究报道HIF-1α是否与心肌缺血后适应有关。本研究通过建立大鼠在体缺血再灌注和缺血后适应模型,探讨HIF-1α对大鼠心肌缺血后适应的心肌保护作用和机制,为心外科手术心肌保护提供新的治疗方向,现报道如下。
SPF级成年SD大鼠40只,雌雄不限,体重300~330 g(辽宁长生生物技术有限公司提供),随机分为4组,每组10只。对照组(A组):仅在冠状动脉前降支穿线,不结扎,持续225 min;缺血再灌注组(B组):阻断前降支45 min,后再灌注3 h;缺血后适应组(C组):阻断前降支45 min后,再灌注开始瞬间实施再灌注10 s-缺血10 s,共3个循环的干预,再灌注3 h;缺血后适应+HIF-1α抑制剂组(D组):阻断前降支45 min后,腹腔注射HIF-1α抑制剂AG490(3 μg/g),再灌注瞬间实施再灌注10 s-缺血10 s,共3个循环的干预,再灌注3 h。
肌酸激酶自动测试盒(奥林巴斯诊断产品有限公司),辣根过氧化物酶标记山羊抗兔IgG(H+L)(碧云天生物技术有限公司),Western二抗稀释液(碧云天生物技术有限公司),Rabbit Anti-GAPDH antibody(博奥森生物有限公司)。
大鼠称重,用5%水合氯醛溶液(0.6 mL/100 g)经腹腔注射麻醉后仰卧位固定于手术台上,将心电图机针形电极插入大鼠四肢皮下记录Ⅱ导联心电图,行气管插管,接小动物呼吸机,呼吸频率60次/min。右股动脉插管取血及补液,自左侧3~4肋间逐层钝性分离皮下、肌肉,打开心包膜暴露左心耳,自左心耳下方2~3 mm处进针,以6-0 prolene缝线在冠状动脉左前降支下穿过,用橡皮管套套锁阻断,阻断缺血45 min后,松开橡皮套再灌注3 h,心电图表现为R波增高、变宽,伴ST段抬高,伴有心律失常,确定大鼠缺血再灌注模型成功建立。
在缺血再灌注模型的基础上,在再灌注开始瞬间控制橡皮套套锁实施再灌注10 s-缺血10 s,共3个循环的干预,随后开放再灌注3 h,建立缺血后适应模型。
分别于术前及心肌缺血45 min、再灌注3 h后3个时间点取血测定血清肌酸激酶的含量。
处死大鼠后取下心脏,剪去心房及右心室,留取左心室切片,于甲醛中固定后在实体显微镜下放大、拍照,导入电脑,用图像分析系统计算出梗死面积占总面积的百分比。四唑(2,3,5-Triphenyl tetrazolium chloride,TTC)染色法梗死区呈白色,未梗死心肌染为红色,分别测量红色、白色区域心肌的面积。
细胞裂解法提取各组细胞总蛋白,BCA法检测蛋白质浓度,按照BCA蛋白浓度测定试剂盒(碧云天生物技术有限公司)的使用说明测定蛋白浓度。将样品蛋白浓度调为一致,每孔上样量为80 μg蛋白。连接电泳装置,打开电源开关,调节电压至40 V,以恒电压方式进行电泳。进行转膜、封闭、抗体免疫杂交,最后进行化学发光(ECL)与曝光。通过灰度分析软件测定灰度(OD)值代表相应蛋白表达量。
采用SPSS 13.0统计软件,计量资料以均数±标准差(Mean±SD)表示,组间比较采用单因素方差分析,多个样本均数的两两比较采用LSD -t检验,P<0.05为差异有统计学意义。
A组大鼠在正常开胸状态下,仅偶尔出现室性早搏(图1A),B组结扎后及再灌注时出现以下变化(图1B、图1C、图1D):结扎冠状动脉后,R波增高、变宽,伴ST段抬高。再灌注后表现为增高的R波振幅回落,抬高的ST段下降。
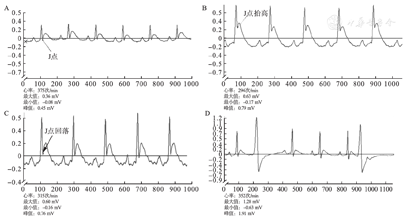
A.A组大鼠正常心电图;B.B组大鼠冠状动脉结扎后QRS波幅增高,ST段显著抬高;C.ST段相对降低(>50%);D.室性早搏。
4组大鼠血清肌酸激酶含量 在结扎前(术前)差异无统计学意义;缺血45 min后,B组、C组以及D组与A组比较差异有统计学意义(P<0.01);再灌注3 h后,B组、C组以及D组与A组比较差异均有统计学意义(P均<0.01),且B组、D组显著高于C组(P<0.05),但此两组比较差异无统计学意义(P>0.05)。见表1。
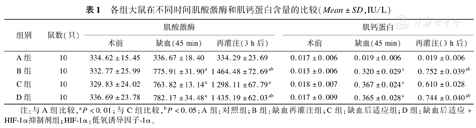
各组大鼠在不同时间肌酸激酶和肌钙蛋白含量的比较(Mean±SD,IU/L)
各组大鼠在不同时间肌酸激酶和肌钙蛋白含量的比较(Mean±SD,IU/L)
| 组别 | 鼠数(只) | 肌酸激酶 | 肌钙蛋白 | ||||
|---|---|---|---|---|---|---|---|
| 术前 | 缺血(45 min) | 再灌注(3 h后) | 术前 | 缺血(45 min) | 再灌注(3 h后) | ||
| A组 | 10 | 334.62±15.45 | 336.67±18.40 | 334.29±23.69 | 0.017±0.006 | 0.019±0.006 | 0.019±0.006 |
| B组 | 10 | 332.77±25.99 | 775.91±31.90a | 1 464.48±72.69ab | 0.015±0.006 | 0.320±0.029a | 0.752±0.039ab |
| C组 | 10 | 329.83±24.02 | 763.82±13.14a | 1 298.11±67.79a | 0.018±0.007 | 0.367±0.024a | 0.610±0.028 |
| D组 | 10 | 336.69±23.78 | 782.17±34.48a | 1 435.19±62.03ab | 0.017±0.009 | 0.365±0.028a | 0.744±0.040ab |
注:与A组比较,aP<0.01;与C组比较,bP<0.05;A组:对照组;B组:缺血再灌注组;C组:缺血后适应组;D组:缺血后适应+HIF-1α抑制剂组;HIF-1α:低氧诱导因子-1α。
4组大鼠血清肌钙蛋白含量 在结扎前(术前)差异无统计学意义;缺血45 min后,B组、C组以及D组肌钙蛋白均高于A组(P<0.01),而3组间比较差异无统计学意义(P>0.05);再灌注3 h后,B组、C组及D组肌钙蛋白与A组比较差异均有统计学意义(P均<0.01),且B组、D组显著高于C组(P<0.05),但此两组比较差异无统计学意义(P>0.05)。见表1。
肉眼观察B组、C组及D组梗死区较A组变薄、暗淡苍白、室壁塌陷,见图2A、图2B。经TTC染色后,肉眼可见A组因未见明显心肌梗死,心脏切片呈红色(图3A),而B组、C组及D组心肌梗死病灶明显,梗死区因脱氢酶流失而呈白色(图3B)。
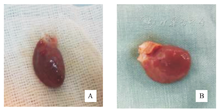
A.缺血再灌注组、缺血后适应组及缺血后适应+HIF-1α抑制剂组心脏暗灰色或青紫色;B.对照组大鼠心脏红润有光泽。

A.对照组心肌染色全部为砖红色;B.缺血再灌注组、缺血后适应组及缺血后适应+HIF-1α抑制剂组心肌染色可见梗死区为苍白色;HIF-1α:低氧诱导因子-1α。
B组[(45.81±5.96)%]、C组[(37.17±4.99)%]及D组[(45.00±3.29)%]与A组[(2.46±1.13)%]心肌梗死面积比较,差异均有统计学意义(P<0.01);B组、D组与C组心肌梗死面积比较,差异有统计学意义(P<0.05)。
C组HIF-1α蛋白的表达最多,其次为B组以及A组,而D组心肌HIF-1α蛋白几乎不表达,各组间比较差异有统计学意义(P<0.05)。见图4。
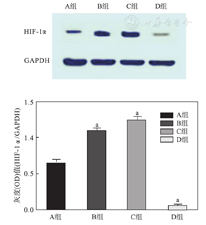
A.对照组;B.缺血再灌注组;C.缺血后适应组;D.缺血后适应+HIF-1α抑制剂组;HIF-1α:低氧诱导因子-1α;GAPDH:内参;与对照组比较,aP<0.05。
长久以来科学家们一直在寻找不同的方法减轻缺血再灌注损伤,更好地实现心肌保护。有研究缺血后适应对心肌缺血再灌注损伤保护作用的机制,发现此干预是由多个因素参与的复杂过程[5,6],其中包括抑制线粒体内钙超载、缓解内皮功能失调、降低氧自由基产生,以及与激活再灌注损伤途径相关,也包括众多的蛋白激酶途径[7]。由于缺血后适应可控性较缺血预适应有更高的可操作性,并且由于其减少了氧自由基的产生,因此有更大的临床可行性和更广的应用前景。
临床上,血清肌酸激酶和肌钙蛋白的脏器特异性升高与是否溶血无关,且其变化可以迅速反映病变的严重程度和病灶范围的大小,假阳性少见而广泛用于急性心肌梗死患者的诊断,两者在骨骼肌和心肌细胞的细胞质中含量极为丰富,脑组织和平滑肌细胞中的含量仅次于骨骼肌细胞[8]。心肌梗死后肌酸激酶和肌钙蛋白易于被检测的原因是因为细胞膜的通透性增高,它们能从细胞内溢出入血[9]。因此,我们在SD大鼠心肌缺血再灌注/心肌缺血后适应模型中选择这两个指标作为评价心肌损伤的标准。心肌梗死面积大小亦是反映心肌损伤程度较好的指标。
对比对照组,本研究发现缺血再灌注可以导致肌酸激酶、肌钙蛋白和心肌梗死面积都有上升趋势,且差异有统计学意义。在缺血再灌注基础上给予后适应干预能显著降低三项指标,说明后适应干预对心肌缺血再灌注具有明显的保护作用。此外,通过采用蛋白印迹试验检测心肌组织中HIF-1α表达的水平,结果提示HIF-1α蛋白在对照组的非缺氧心肌中表达水平较低,在缺血再灌注组、缺血后适应组急剧增加,而在缺血后适应+HIF-1α抑制剂组几乎不表达。此结果说明缺血再灌注增加了HIF-1α的表达,是细胞对这一人为干预做出的适应性反应。且HIF-1α在缺血后适应组中的表达较缺血再灌注组更加显著,说明缺血后适应能够增加HIF-1α的表达,提示HIF-1α可能参与缺血后适应心肌保护作用的过程。研究表明,缺血预适应能够诱导HIF-1α蛋白表达的增加[10,11]。HIF-1α受细胞内氧浓度的影响大,能较快地在分子水平上做出反应。本实验结果提示HIF-1α在缺血后适应的干预后表达明显增加。因此,我们推测HIF-1α表达增加对缺血后适应的干预同样存在心肌保护作用,是实现缺血后适应心肌保护作用的一个重要转录因子。
20世纪90年代初,Samelko等[12]在低氧的Hep3B肝癌细胞株的细胞核中发现一种转录因子,在低氧条件下能特异性地结合促红细胞生成素基因上的缺氧反应元件(hypoxia response ele-ments,HRE),促使该因子的转录,被命名为HIF-1。
在常氧条件下,HIF-1α正常表达,但被迅速的泛素化。在氧分子、铁离子、α-酮戊二酸的参与下,其氧依赖降解结构域(the oxygen-dependent degradation domain,ODD)上的脯氨酸残基能被脯氨酰羟化酶所修饰,然后立即与希佩尔-林道肿瘤抑制蛋白(Von Hippel-Lindau tumor suppressor protein,pVHL)结合,pVHL与ODD结合后作为E3泛素化连接酶复合物的识别组件,介导HIF-1α蛋白的降解,以上所述的酶都是氧依赖的[13]。抑制HIF-1α的转录活性被认为是除上述方式以外的又一氧依赖调节机制。在缺氧环境下,HIF-1从细胞质转移至细胞核,α与β亚基形成的异二聚体变得非常稳定,HIF-1α的核易位避免遭受pVHL介导蛋白降解[14]。因此,缺氧状态的持续存在对HIF-1α的稳定性起至关重要的作用。
分子水平的转录过程是由转录因子与特定的靶基因序列结合实现的,同样HIF-1α亚基的转录活性由靶基因上的HRE数量和与其结合的难易程度决定[15]。HIF-1α作为一种核转录因子,是维持体内稳态平衡调节的重要因子,其调节作用与哺乳动物正常发育密切相关,在病理生理反应过程中发挥至关重要的作用。至今已发现有100多个下游靶基因受HIF-1调控[16],如调节红细胞生成、促进血管新生、参与调控细胞增殖与凋亡,调节葡萄糖和能量代谢,维持组织及细胞在低氧条件下内环境的稳定等[17]。
21世纪初,学者们逐渐发现HIF-1α与促红细胞生成素、血红素氧合酶1(heme oxygenase 1,HO-1)、诱导型一氧化氮合酶(inducible nitric-oxide synthase, iNOS)以及血管内皮生长因子(vascular endothelial growth factor,VEGF)等诱导发挥心肌保护作用的分子之间有紧密联系[18,19,20,21]。HO-1参与血红素代谢的关键步骤,血红素分解代谢生成一氧化碳(CO),CO能插入通道蛋白胞浆中的C末端实现对大鼠心肌L型Ca2+通道和人心肌L型Ca2+通道a1c亚单位的直接抑制[22]。并且这种非电压依赖的通道抑制是可逆的,而HO-1是HIF-1α的靶基因,HIF-1α对其诱导可能是一个心肌保护机制[23]。此外,HIF-1α可以通过促进促红细胞生成素表达激活P38信号上调HO-1实现间接心肌保护作用;Cai等[24]发现,短暂低氧环境可引起iNOS表达增加,并减少了心肌坏死程度、减轻了因缺血导致的心功能不全。通过进一步的研究发现,iNOS基因在其启动子区含有HRE,因此推测此作用是由于HIF-1α与iNOS基因启动子区的HRE相结合发挥的心肌保护作用;Kido等[25]建立转基因小鼠的急性心肌缺血模型发现,HIF-1α、VEGF表达上调,且VEGF迟于HIF-1α mRNA出现,伴心肌毛细血管密度增加;而对照组心肌标本并无HIF-1α和VEGF mRNA的表达,表明HIF-1α能调控VEGF的表达。这些转录因子均减轻了心肌再灌注损伤。
有实验证明低氧诱导因子基因被敲除的小鼠中,经过缺血预处理后未显示出心肌保护作用[26],且当遭受致命性缺血损伤时心肌梗死面积显著增大、心功能急剧恶化[27],而经缺血预处理的野生型小鼠中,再灌注后显现出较对照组更高的射血分数和更小的梗死面积,因此提出HIF在细胞中的持续表达可以抵抗再灌注损伤,且这一作用与该因子数量有关[28]。
Eckle等[29]发现HIF-1α增强嘌呤途径介导缺血预适应,A2B亚型的腺苷受体参与这一过程;Zhao等[30]在正常和高胆固醇血症的动物中发现,HIF的上调与心肌保护作用关系密切。既然HIF-1α在缺血预适应引发的心肌保护中均发挥重要作用,预适应中激活了众多信号通路,且均与HIF-1α有关,同时该因子调控着许多靶基因实现心肌保护作用,那缺血后适应引起的心肌保护作用是否亦与HIF-1α有关?为了验证我们的想法,本实验中使用了HIF-1α的抑制剂AG490抑制蛋白的表达。
本实验在大鼠在体心肌缺血再灌注损伤模型的基础上建立缺血后适应模型,检测各时间点的血清肌酸激酶、肌钙蛋白含量,测定心肌梗死面积,检测HIF-1α蛋白含量,发现缺血后适应可以减轻心肌因缺血再灌注导致的进一步损伤,心肌梗死面积、血清肌酸激酶、肌钙蛋白含量均较急性心肌缺血再灌注降低,实现了心肌保护作用。同时给予AG490(HIF-1α抑制剂)抑制HIF-1α在蛋白水平的表达,发现心肌梗死面积增加,血清中肌酸激酶含量升高,从反面证明缺血后适应对心肌的保护作用。
本研究结果显示,与缺血后适应组相比,缺血后适应+HIF-1α抑制剂组的HIF-1α蛋白水平表达几乎为零。同时,较缺血后适应组,HIF-1α抑制剂组心肌梗死面积增大,血清中肌酸激酶、肌钙蛋白含量升高,提示HIF-1α抑制剂与缺血后适应的心肌保护作用具有拮抗效应。从反面表明,经过后适应干预HIF-1α表达上调,实现了减轻再灌注损伤的作用。
综上所述,多次反复短暂的缺血再灌注形成了短暂缺氧的环境,上调了HIF-1α的表达,实现了保护心肌的作用。本研究结果为体外循环手术后心肌缺血再灌注损伤提供了新的治疗方案。
所有作者均声明不存在利益冲突

























