
探讨新生儿先天性高胰岛素血症(CHI)的临床特点、治疗、预后及基因突变情况。
选取河北省儿童医院新生儿重症监护室2017年2月至2020年8月收治的CHI患儿为研究对象,对其临床特点、诊治、预后及基因检测结果进行回顾性分析。
共纳入7例CHI患儿,平均胎龄(38.1±1.5)周,2例为早产儿(胎龄<37周),平均出生体重(3 608±906)g,3例为巨大儿(出生体重>4 000 g)。主要临床表现包括嗜睡、吃奶差、肤色发绀、抽搐、四肢抖动等,2例早产儿无特异性表现,分别于生后1 h、3 h常规监测血糖时发现明显异常。7例患儿中,6例从入院就需要较高的葡萄糖输注速率[>10 mg/(kg·min)]以维持血清葡萄糖的正常水平,只有1例入院初期葡萄糖输注速率仅需3~5 mg/(kg·min),后期最高升至8 mg/(kg·min)。7例患儿均给予二氮嗪口服,2例(2/7)有效,其中1例于6月龄停药,监测血糖正常。其余5例(5/7)患儿由于编码胰岛β细胞KATP+通道的ABCC8基因突变而对二氮嗪耐药,给予奥曲肽治疗。其中2例(2/5)对奥曲肽治疗有效;3例(3/5)对二氮嗪和奥曲肽治疗均无效。随访7例患儿中,有3例体格及神经系统发育未见异常,2例体格发育未见异常,但分别有癫痫及语言、个人-社交中度发育迟缓。另2例患儿1例死亡,1例失访。基因检测结果显示,6例为ABCC8基因杂合突变,1例为GLUDI基因杂合突变。
CHI的临床表现缺乏特异性,对存在高危因素的患儿应及时完善血糖监测。对于葡萄糖输注速率不高[<8 mg/(kg·min)]的患儿不能忽视CHI。有些患儿可自行缓解,分子诊断可明确致病基因,早期识别、规范管理可改善患儿预后。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性高胰岛素血症(congenital hyperinsulinism,CHI)是一种罕见疾病,其特征是胰岛素释放不受调节,导致低血糖,是新生儿期和儿童早期持续和严重低血糖的最常见原因。及时诊断和即刻处理低血糖对于避免低血糖性脑损伤和长期的神经系统并发症至关重要[1]。基因检测技术是明确诊断CHI的重要手段,目前已知至少16个关键基因突变与CHI发病有关[2],明确基因突变类型对CHI的诊疗和预后评估有重要意义。本研究通过对7例新生儿CHI的临床资料、基因突变及预后进行分析,旨在了解CHI基因突变类型与临床表型及预后的关系。
选择2017年2月至2020年8月我院新生儿重症监护室收治的经基因检查确诊为CHI的7例患儿进行回顾性分析。研究内容包括:患儿的性别、日龄、出生胎龄、出生体重、发病日龄等人口学特征、临床表现、接受治疗情况、实验室检查、基因检查结果以及预后。
依据第5版《实用新生儿学》[3],符合以下6项:(1)新生儿期反复低血糖发作,多为严重低血糖,甚至<1 mmol/L或不能测出;(2)绝对或相对持续高胰岛素血症,如低血糖时空腹血胰岛素>10 IU/L;血糖0.6~0.8 mmol/L时,血胰岛素>5 IU/L;血胰岛素(IU/L)/血葡萄糖(mg/dL)比值>0.3等均提示高胰岛素血症;(3)低血糖时无酮症;(4)静脉注射葡萄糖需要≥8 mg/(kg·min)以上才能维持血糖正常;(5)一般影像学检查无异常发现;(6)其他:胰岛素对抗类激素水平如皮质醇水平未见增高,或使用激素、胰高血糖素、奥曲肽等诊断性治疗时能有效提高血糖水平等均提示或证实CHI的诊断。
从受检者及其父母外周血中提取基因组DNA,经片段化、连接接头、扩增纯化后,使用Fulgent WES(IDT)杂交捕获人类全部基因20 099的外显子区及相邻内含子区域(50 bp),捕获到的DNA经洗脱和扩增纯化后,使用高通量测序仪(Illumina NovaSeq6000)进行测序。
应用SPSS 16.0软件进行统计学分析,符合正态分布的计量资料以均数±标准差(Mean±SD)表示,计数资料以例数表示。
7例CHI患儿中,男4例,女3例,足月儿5例,早产儿2例,胎龄36.0~40.3周,平均胎龄(38.1±1.5)周,出生体重2 300~4 880 g,平均出生体重(3 608±906)g,3例患儿出生体重>4 000 g。起病时间为生后14 min~24 h,最低血糖为0.4~1.4 mmol/L,5例患儿母亲均否认糖尿病及低血糖病史,病例3患儿母亲患妊娠期糖尿病,病例7患儿母亲患1型糖尿病,病例6患儿姐姐存在ABCC8基因位点突变。
7例CHI患儿的临床表现、生化检查结果及干预效果见表1。7例患儿中,5例有嗜睡、吃奶差症状,2例早产儿无特异性表现,分别于生后1 h、3 h常规监测血糖时发现明显异常。所有患儿均在入院后接受葡萄糖静推,持续静点葡萄糖,并静点氢化可的松琥珀酸钠提升血糖,7例患儿中,6例从入院就需要较高的葡萄糖输注速率[>10 mg/(kg·min)]以维持血清葡萄糖的正常水平,只有1例入院初期葡萄糖输注速率仅需3~5 mg/(kg·min),后期最高升至8 mg/(kg·min)。诊断高胰岛素血症后,7例患儿口服二氮嗪,起始剂量为5 mg/(kg·d),根据患儿病情逐渐增加剂量,最大剂量为15 mg/(kg·d),其中2例患儿(病例1和4)对二氮嗪治疗有效,其中1例已停药,监测血糖正常。二氮嗪治疗无效的5例患儿改为奥曲肽治疗,奥曲肽起始剂量为5 μg/(kg·d),皮下注射,根据病情逐渐增加剂量,最大剂量为25 μg/(kg·d),其中2例对奥曲肽有效(病例3和7),另外3例对二氮嗪及奥曲肽均无效,见表1。病例2就诊于上海某医院,行PET-CT检查提示胰腺18氟-多巴放射性摄取弥漫性增高,行胰腺大部分切除术;病例5就诊于上海某医院,行PET-CT检查提示胰腺18氟-多巴代谢弥漫性增高,家属未行手术,放弃治疗;病例6失访。
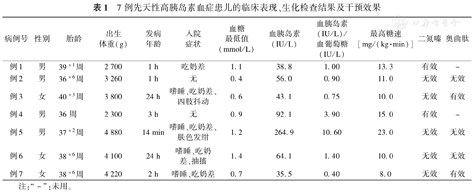
7例先天性高胰岛素血症患儿的临床表现、生化检查结果及干预效果
7例先天性高胰岛素血症患儿的临床表现、生化检查结果及干预效果
| 病例号 | 性别 | 胎龄 | 出生体重(g) | 发病年龄 | 入院症状 | 血糖最低值(mmol/L) | 血胰岛素(IU/L) | 血胰岛素(IU/L)/血葡萄糖(IU/L) | 最高糖速[mg/(kg·min)] | 二氮嗪 | 奥曲肽 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 例1 | 男 | 39+1周 | 2 700 | 1 h | 吃奶差 | 1.1 | 38.8 | 1.00 | 13.3 | 有效 | - |
| 例2 | 男 | 36+6周 | 3 260 | 1 h | 无 | 0.4 | 56.0 | 0.90 | 11.0 | 无效 | 无效 |
| 例3 | 女 | 40+3周 | 3 800 | 24 h | 嗜睡、吃奶差、四肢抖动 | 0.6 | 43.1 | 0.75 | 10.0 | 无效 | 有效 |
| 例4 | 男 | 36周 | 2 300 | 3 h | 无 | 0.9 | 92.1 | 3.90 | 15.0 | 有效 | - |
| 例5 | 男 | 37+2周 | 4 880 | 14 min | 嗜睡、吃奶差、肤色发绀 | 1.2 | 264.9 | 10.60 | 23.0 | 无效 | 无效 |
| 例6 | 女 | 38+6周 | 4 100 | 24 h | 嗜睡、吃奶差、抽搐 | 1.4 | 64.1 | 1.40 | 10.0 | 无效 | 无效 |
| 例7 | 女 | 38+6周 | 4 220 | 2 h | 嗜睡、吃奶差 | 0.7 | 35.5 | 0.40 | 8.0 | 无效 | 有效 |
注:"-":未用。
7例患儿的基因检测结果见表2,6例为ABCC8基因杂合突变,表现为多位点突变,1例为GLUDI基因突变,7例患儿中2例为复合杂合突变,余5例为杂合突变。
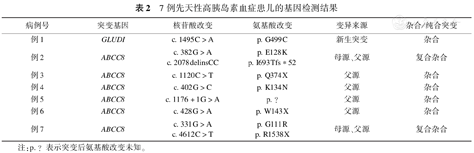
7例先天性高胰岛素血症患儿的基因检测结果
7例先天性高胰岛素血症患儿的基因检测结果
| 病例号 | 突变基因 | 核苷酸改变 | 氨基酸改变 | 变异来源 | 杂合/纯合突变 |
|---|---|---|---|---|---|
| 例1 | GLUDI | c.1495C>A | p.G499C | 新生突变 | 杂合 |
| 例2 | ABCC8 | c.382G>Ac.2078delinsCC | p.E128Kp.I693Tfs*52 | 母源、父源 | 复合杂合 |
| 例3 | ABCC8 | c.1120C>T | p.Q374X | 父源 | 杂合 |
| 例4 | ABCC8 | c.402G>C | p.K134N | 父源 | 杂合 |
| 例5 | ABCC8 | c.1176+1G>A | p.? | 父源 | 杂合 |
| 例6 | ABCC8 | c.428G>A | p.W143X | 父源 | 杂合 |
| 例7 | ABCC8 | c.331G>Ac.4612C>T | p.G111Rp.R1538X | 母源、父源 | 复合杂合 |
注:p.?表示突变后氨基酸改变未知。
病例1患儿(GLUDI基因突变)对二氮嗪治疗有效,长期服用二氮嗪,监测血糖正常,生后5岁1个月评估,体格发育正常,但患儿患癫痫,动态脑电图提示背景活动慢波增多,广泛性棘波、多棘慢波、多棘波发放,需长期服用抗癫痫药物治疗。
其余6例患儿均为ABCC8基因突变,对药物治疗效果及预后存在较大差异。病例4患儿应用二氮嗪治疗有效,服药6个月后停药,监测血糖正常,生后2岁4个月评估,体格及神经系统发育未见异常。病例3及病例7患儿对二氮嗪无效,对奥曲肽有效,现奥曲肽皮下注射,血糖监测正常,2例患儿有胆结石及肝功能损害等不良反应,生后2岁3个月、1岁7个月评估,体格及神经系统发育未见异常。病例2、5、6患儿对二氮嗪及奥曲肽均无效,病例2行胰腺大部分切除术,目前糖奶喂养(奶粉添加麦芽糊精)情况下监测血糖正常,生后2岁11个月评估体格发育未见异常,适应性、大运动、精细动作轻度发育迟缓,语言、个人-社交中度发育迟缓。病例5患儿生后40 d行PET-CT检查提示胰腺18氟-多巴代谢弥漫性增高,患儿出现心力衰竭,家属拒绝手术、放弃治疗后死亡。病例6患儿失访,家属拒绝提供预后资料。
CHI是一组具有遗传异质性和临床表现异质性的综合征,迄今已发现16种致病基因与之相关,包括ABCC8、KCNJ11、GLUD1、GCK、HADH、SLC16A1、UCP2、HNF4A、HNF1A、HK1、KCNQ1、CACNA1D、FOXA2、EIF2S3、PGM1和PMM2[2],但仍有约一半的CHI患儿未能确定潜在的遗传原因[4]。另外,还有以综合征形式存在的高胰岛素血症,包括先天性糖基化障碍、Kabuki综合征、Beckwith-Wiedemann综合征、Turner综合征等[5]。CHI可以为暂时性,也可以是永久性的,在疾病初期难以区分,因此,对持续或严重低血糖患儿需尽早完善基因检测,指导临床治疗及判断预后。
人群中CHI的发病率为1∶3.5万~1∶4万[6],英国的最近一项研究表明,每28 389例活产儿中至少有1例CHI[7],目前我国尚无CHI在人群中的发病率报道。国内Wang等[8]报道2017年之前CHI病例绝大多数来自北京市、上海市以及广东省、浙江省等,国内其他地区报道很少。我院的7例病例均系在2017年后确诊,在此之前因对该病认识不足,基因检测技术未广泛开展,可能存在本地区CHI病例漏诊,也有部分病例转至省外治疗,确诊后未做报道。
新生儿CHI早期症状不典型,可表现为喂养困难、嗜睡、吃奶差、多汗、肌张力低下、呼吸暂停等,症状重者可能出现危及生命的神经系统症状如抽搐、意识丧失、昏迷,甚至死亡[9]。本组病例中有5例早期出现嗜睡、吃奶差等非特异性临床表现,另2例早产儿无低血糖症状,均系生后早期常规监测血糖时发现明显异常。因此,早期识别低血糖高危因素,对胎龄<37周、小于胎龄儿、大于胎龄儿、母亲糖尿病、产时窒息、红细胞增多症、溶血性贫血等高危因素的患儿进行常规血糖监测,对于减少漏诊至关重要[10]。
CHI的治疗手段主要包括急性期输注葡萄糖及频繁喂养、长期内科药物治疗(主要包括二氮嗪、生长抑素类似物等)以及外科手术治疗[11]。本组病例中所有患儿均给予葡萄糖输注及应用二氮嗪治疗,5例二氮嗪治疗无效的患儿应用奥曲肽治疗,只有病例2行手术治疗。低血糖往往需要较高的葡萄糖输注速率才能维持血糖在正常范围,静脉注射葡萄糖速率8 mg/(kg·min)以上才能维持血糖正常是CHI的诊断指标之一[12],但并非所有病例均符合。本研究中病例7生后2 h即发现低血糖,于出生医院新生儿科治疗3 d后出院,院外出现嗜睡、吃奶差,测血糖明显低于正常,就诊于我院,住院初期葡萄糖输注速率仅需3~5 mg/(kg·min)即可维持血糖正常,后期最高升至8 mg/(kg·min)。该例患儿的特点是入院初期所需葡萄糖输注速率并不高,但不能耐受速率下调,需持续静点葡萄糖。此类患儿容易被临床医生忽视,导致出院过早,再加上随访策略不规范,增加患儿遗留神经系统后遗症的风险。临床工作中需要规范血糖监测流程,对于不能耐受减量或停用静脉输注葡萄糖的患儿要高度警惕,及时完善基因检测。
CHI最常见、最严重的形式是ABCC8和KCNJ11基因的失活突变,占所有CHI病例的40%~50%[13]。本研究显示,同是ABCC8基因突变的患儿,对药物敏感性存在差异。ABCC8和KCNJ11分别编码胰腺β细胞ATP敏感性钾通道的两个亚基(分别为SUR1和Kir6.2),基因突变导致其编码的SUR1及Kir6.2失活,使KATP+通道持续关闭,细胞膜持续去极化,导致胰岛素分泌失调。该类型的CHI遗传方式为常染色体隐性或显性遗传。二氮嗪为KATP+通道拟似剂,遗传方式为常染色体隐性遗传的患儿,由于完全缺乏KATP+通道,因此对二氮嗪治疗无反应。相比之下,显性遗传的患儿存在不同程度的通道活性受损,严重损害者对二氮嗪治疗无反应,而部分损害者具有残留KATP+活性,对二氮嗪治疗有反应。文献报道,ABCC8基因突变位点较多,对二氮嗪的总体疗效欠佳[4,14]。本研究也发现,6例(85.7%)患儿存在ABCC8突变,其中5例突变位点曾被报道,对二氮嗪治疗均无效。
GLUD1基因突变引起的高胰岛素血症/高氨血症综合征为CHI的第2类常见原因[1],由编码谷氨酸脱氢酶的GLUD1突变引起,禁食或进食富含蛋白质的食物后会出现反复低血糖发作,此基因突变所致CHI通常病情较轻,出生时非巨大儿,通常要到儿童或成人后期才能确诊[15]。Roy等[16]报道3例CHI患儿发病年龄为5个月~5岁,Stanley等[17]报道8例确诊CHI患儿的年龄为3个月~10岁。这可能是由于与β细胞KATP+通道缺陷者相比,这些患儿生后早期低血糖症的严重程度较低,且富含蛋白质的固体食物通常在出生几个月后才会被逐渐引入,因此确诊时间较晚。此类CHI虽对二氮嗪和(或)饮食管理反应良好,但部分患儿由于诊断延迟、二氮嗪成本较高和受药物供应限制,神经系统后遗症比较常见[16,18,19]。本研究中病例1系在新生儿期即确诊,目前口服二氮嗪情况下监测血糖正常,但仍需应用抗癫痫药物治疗,远期神经系统预后需继续随访。
存在基因突变的CHI患儿中,部分可经治疗后病情缓解,有研究报道32例CHI患儿中有10例在12个月内缓解,其中包括3例父系遗传的KCNJ11突变患儿,不需要行胰腺切除手术[20]。英国Kumaran等[21]报道1例CHI新生儿,发生ABCC8复合杂合突变(R168C/S606T),生后8周内高胰岛素血症、低血糖完全缓解。另有1例高胰岛素血症/高氨血症综合征患儿,基因分析证实GLUD1突变,8岁时自发缓解,停用二氮嗪[18]。本研究中病例4在6个月内病情完全缓解,停用二氮嗪后血糖及胰岛素水平正常,该患儿系父系遗传的ABCC8基因突变(c.402G>C),此突变位点为HGMD未收录的变异,归类为意义未知的变异,需进一步完善功能试验。目前,许多新发突变的自然病程尚属未知,CHI可能自发缓解,临床医生应反复权衡,避免盲目的早期胰腺手术,同时保证维持血糖在正常水平,并为家长提供相关预后资料。
总之,早期识别低血糖高危因素,及时识别高胰岛素血症,对避免严重低血糖相关脑损伤和远期神经系统后遗症至关重要。对于所需葡萄糖输注速率不高的患儿也不能忽视,有些病例可自行缓解。分子诊断可明确致病基因,有助于制定规范化随访策略及判断预后,使CHI患儿得到更精准治疗并提升生存质量、改善预后,并对下一代的发病风险提供遗传咨询。
所有作者均声明不存在利益冲突

























