
比较不同方法提取真菌基因组DNA的优劣,寻找一种用于PCR扩增的快速、高效的真菌DNA提取方法。
分别用加热裂解法、微波法、反复冻融法、溶壁酶法、蜗牛酶法和试剂盒法提取浙江中医药大学附属杭州第一医院保存的马尔尼菲青霉、小孢根霉、新型隐球菌和白假丝酵母菌DNA,在凝胶成像系统下观察基因组DNA电泳图。采用超微量核酸蛋白测定仪测定基因组DNA的浓度和纯度,并计算产率;同时对提取的基因组DNA进行PCR扩增并测序。采用方差分析和SNK–q检验比较不同方法所得基因组DNA浓度和产率。
6种方法提取马尔尼菲青霉(霉菌相和酵母相)、小孢根霉、新型隐球菌和白假丝酵母菌所得DNA浓度和产率差异均存在统计学意义(F=750.83,220.95,669.35,132.01,510.20和1658.35,287.10,963.64,1147.77,4521.22,P值均<0.01),且微波法所得真菌DNA浓度和产率最高,加热裂解法次之,试剂盒法最低。所有方法所得DNA经PCR扩增均出现阳性条带。
6种方法均可成功提取真菌基因组DNA,微波法和加热裂解法操作简便,所得真菌基因组DNA模板能够满足临床实验室PCR扩增需求。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
随着广谱抗生素、免疫抑制剂和激素类药物等的广泛使用以及HIV感染率的上升,侵袭性真菌病(Invasive fungal disease, IFD)的高发病率与病死率已引起了人们的广泛关注,早期、快速诊断和治疗对提高IFD的治愈率和患者的生存质量意义重大[1]。传统的真菌鉴定多以形态学为依据,存在不确定性,对于少见致病真菌诊断困难。随着分子生物学技术的发展,真菌鉴定可至种的水平。目前实验室对于难鉴定的真菌多采用PCR扩增结合测序分析的方法,该法具有准确、简单、快速、实用性强的特点,而进行PCR扩增首先需要提取真菌基因组DNA,这时探索出一种快速、高效、安全的真菌基因组DNA抽提法显得尤为重要。本实验采用微波法、加热裂解法、反复冻融法、试剂盒法、溶壁酶法和蜗牛酶法,试图寻找一种真菌基因组DNA快速提取方法,为临床应用PCR快速高效地鉴定真菌奠定基础。
选用的菌株为杭州市第一人民医院检验科保存的白假丝酵母菌、新型隐球菌、马尔尼菲青霉和小孢根霉,均分离自患者,其中新型隐球菌、马尔尼菲青霉和小孢根霉经DNA测序鉴定,白假丝酵母菌经全自动微生物分析系统Vitek 2 Compact鉴定。
沙保罗培养基、溶壁酶、蜗牛酶由上海源叶生物公司提供,DNA专用提取液由广东达安基因诊断公司提供;真菌DNA提取试剂盒由Qiagen公司提供,ABI 2720 PCR仪器由美国ABI公司提供,DK–8D型电热恒温水槽由上海精密实验设备有限公司提供;电泳仪及凝胶成像系统购于美国Bio–Rad公司,Heraeus高速冷冻离心机由德国Sorvall提供,NanoDrop ND–2000/2000C超微量核酸蛋白测定仪由德国Thermo公司提供。
将上述菌种接种于沙保罗培养基上,37℃培养(其中马尔尼菲青霉分别置于25℃和37℃下培养)。白假丝酵母菌和小孢根霉培养1 d,新型隐球菌培养3 d,两种环境培养下的马尔尼菲青霉培养5 d后挑取菌落,加入已经称重的含3 mL无菌等渗氯化钠溶液试管中,再次称重并调整含真菌试管质量,使得每管含真菌300 mg,振荡混匀后分成30管(每种真菌每种提取方法各5管),每管含真菌约10 mg,4℃ 12 000×g离心10 min去上清液,备用。
向上述备用菌中加100 μL DNA专用提取液,振荡混匀,100℃金属浴10 min,4℃ 12 000×g离心5 min,取全部上清液于–20℃保存备用。
对田永强[3]的方法加以改进,向上述备用菌中加入200 μL DNA专用提取液,振荡混匀,微波高火30 s,连续4次,4℃ 12 000×g离心5 min,取上清液于–20℃保存备用。
向上述备用菌中加入1 mL的无菌等渗氯化钠溶液,置–70℃冰箱中10 min,室温融化,重复冻融3次。4℃ 12 000×g离心10 min后去上清液,加入100 μL DNA专用提取液振荡混匀,金属浴加热5 min,4℃ 12 000×g离心5 min,取上清液于–20℃保存备用。
上述备用菌中加500 μL普通DNA裂解液[5](10 mmol/L Tris–HCl pH 8.0,10 mmol/L NaCl,pH 8.0的50 mmol/L EDTA),加溶壁酶[6](50 mg/mL)10 μL,水浴2 h后。加入等体积苯酚–氯仿–异丙醇(25∶24∶1),轻轻颠倒混淆1 min,4℃ 12 000×g离心5 min。吸取上清液,用等体积氯仿–异丙醇(24∶1)再抽提1次,4℃ 12 000×g离心5 min;取上清液加入2倍体积无水乙醇,–20℃冰箱静置30 min,4℃ 12 000×g离心5 min,弃上清液,加入200 μL 70%乙醇洗涤,4℃ 12 000×g离心5 min,弃上清液,扣干,加入50 μL Tris–EDTA缓冲液中充分溶解,所提DNA溶液于–20℃保存备用。
参照赵宏宇等[7]蜗牛酶过夜处理法,上述备用菌中加500 μL普通DNA裂解液,蜗牛酶(10 mg/mL)100 μL和100 μL 1% SDS,37℃消化14 h,按上述酚氯仿法抽提纯化。
采用Qiagen公司的DNeasy Plant mini kit,按试剂盒说明操作,最后溶于50 μL试剂盒自带洗脱缓冲液中,整个实验过程约耗时50 min。
取上述各种方法提取真菌基因组DNA液各1.5 μL,采用超微量核酸蛋白测定仪测定样品中DNA的浓度和纯度,并根据DNA浓度和每管真菌重量(10 000 μg)之比计算不同提取方法所得真菌基因组DNA的提取产率[9](每μg真菌所得DNA ng数):DNA浓度(ng/μL)×样本体积/10 000 μg,另各取5μL真菌基因组DNA液进行琼脂糖电泳,凝胶成像仪下观察其条带。
将提取的基因组DNA作为模板,以真菌18S rDNA为靶序列(上游引物:5′–GCATCGATGAAGAACGCAGC–3′,下游引物:5′–TCCTCCGCTTATTGATATGC–3′,由上海中科新生命生物科技有限公司合成),按照TaKaRa试剂公司的说明书进行PCR扩增,PCR反应条件为94℃预变性5 min,94℃变性30 s,52℃退火30 s,72℃延伸1 min,循环30次,最后72℃延伸7 min,用1.5%琼脂糖凝胶电泳,在紫外灯检测仪下分析结果并用凝胶图像处理系统照相保存,同时将PCR产物送上海中科新生命生物科技有限公司测序。
采用SPSS 17.0软件进行分析,计量资料采用 ±s表示,采用方差分析,不同提取方法所得DNA浓度和产率差异的两两比较用SNK –q检验。P<0.05为差异有统计学意义。
±s表示,采用方差分析,不同提取方法所得DNA浓度和产率差异的两两比较用SNK –q检验。P<0.05为差异有统计学意义。
真菌基因组DNA提取所需时间由短至长分别为:微波法<加热裂解法<反复冻融法<试剂盒抽提法<溶壁酶法<蜗牛酶法。微波法、加热裂解法和反复冻融法在250 bp处有弥散亮条带,蜗牛酶法和溶壁酶法在大于10×103 bp处可见微弱细条带,试剂盒抽提法未见条带。白假丝酵母菌DNA电泳图见图1。
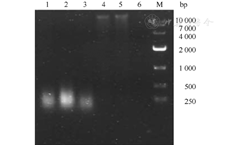
注:M.DNA标准带;1.加热裂解法;2.微波法;3.反复冻融法;4.溶壁酶法;5蜗牛酶法;6试剂盒法
加热裂解法、反复冻融法、微波法、溶壁酶法、蜗牛酶法、试剂盒法的纯度(A260/A280)分别为:1.01±0.11、0.99±0.09、1.06±0.08、1.83±0.18、1.87±0.16和1.70±0.30。
各种方法提取白假丝酵母菌DNA浓度由高至低分别为:微波法>加热裂解法>溶壁酶法>反复冻融法>蜗牛酶法>试剂盒法(P<0.05);新型隐球菌:微波法>加热裂解法>反复冻融法>蜗牛酶法>试剂盒法(P<0.05),加热裂解法和溶壁酶法间差异无统计学意义(P>0.05);马尔尼菲青霉霉菌相:微波法>加热裂解法>溶壁酶法>蜗牛酶法>试剂盒法(P<0.05),加热裂解法和反复冻融法差异无统计学意义(P>0.05);马尔尼菲青霉酵母相:微波法>加热裂解法>反复冻融法>溶壁酶法>试剂盒法(P<0.05),溶壁酶法和蜗牛酶法差异无统计学意义(P>0.05);小孢根霉:微波法>加热裂解法>溶壁酶法>试剂盒法(P<0.05),溶壁酶法和蜗牛酶法差异无统计学意义(P>0.05),加热裂解法和反复冻融法差异无统计学意义(P>0.05)(表1)。
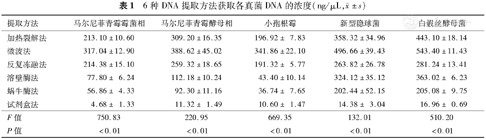
 ±s)±s)
±s)±s)| 提取方法 | 马尔尼菲青霉霉菌相 | 马尔尼菲青霉酵母相 | 小孢根霉 | 新型隐球菌 | 白假丝酵母菌 |
|---|---|---|---|---|---|
| 加热裂解法 | 213.10±10.60 | 309.20±16.35 | 196.92± 7.83 | 358.32±34.96 | 443.10±18.14 |
| 微波法 | 317.04±12.90 | 388.62±45.02 | 341.86±22.10 | 496.66±39.43 | 543.40±11.43 |
| 反复冻融法 | 214.38±15.10 | 259.32±18.65 | 191.32± 5.77 | 263.82±26.78 | 281.24±13.41 |
| 溶壁酶法 | 77.80± 6.24 | 112.18±10.24 | 43.40±10.14 | 324.12±35.12 | 363.02± 6.23 |
| 蜗牛酶法 | 56.86± 4.33 | 92.30±11.16 | 36.74± 7.65 | 202.44±52.15 | 205.08± 9.75 |
| 试剂盒法 | 4.68± 1.33 | 11.32± 1.49 | 10.60± 1.47 | 14.38± 3.04 | 16.96± 0.69 |
| F值 | 750.83 | 220.95 | 669.35 | 132.01 | 510.20 |
| P值 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
各种方法提取白假丝酵母菌、新型隐球菌DNA产率由高至低分别为:微波法>加热裂解法>反复冻融法>溶壁酶法>蜗牛法>试剂盒法(P<0.05);马尔尼菲青霉霉菌相、酵母相:微波法>加热裂解法>溶壁酶法>试剂盒法(P<0.05),加热裂解法和反复冻融法差异无统计学意义(P>0.05);溶壁酶法和蜗牛酶法差异无统计学意义(P>0.05);小孢根霉:微波法>加热裂解法>溶壁酶法(P<0.05),加热裂解法和反复冻融法差异无统计学意义,溶壁酶法和蜗牛酶法和试剂盒法差异无统计学意义(P>0.05)(表2)。
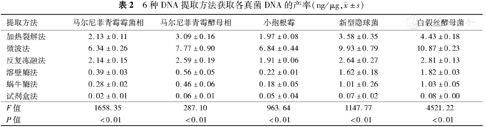
 ±s)±s)
±s)±s)| 提取方法 | 马尔尼菲青霉霉菌相 | 马尔尼菲青霉酵母相 | 小孢根霉 | 新型隐球菌 | 白假丝酵母菌 |
|---|---|---|---|---|---|
| 加热裂解法 | 2.13±0.11 | 3.09±0.16 | 1.97±0.08 | 3.58±0.35 | 4.43±0.18 |
| 微波法 | 6.34±0.26 | 7.77±0.90 | 6.84±0.44 | 9.93±0.79 | 10.87±0.23 |
| 反复冻融法 | 2.14±0.15 | 2.59±0.19 | 1.91±0.06 | 2.64±0.27 | 2.81±0.13 |
| 溶壁酶法 | 0.39±0.03 | 0.56±0.05 | 0.22±0.01 | 1.62±0.18 | 1.82±0.03 |
| 蜗牛酶法 | 0.28±0.02 | 0.46±0.06 | 0.18±0.05 | 1.01±0.26 | 1.03±0.05 |
| 试剂盒法 | 0.02±0.01 | 0.06±0.01 | 0.05±0.04 | 0.07±0.02 | 0.08±0.00 |
| F值 | 1658.35 | 287.10 | 963.64 | 1147.77 | 4521.22 |
| P值 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
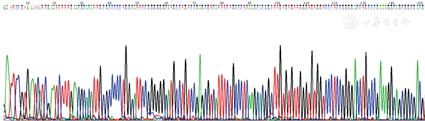
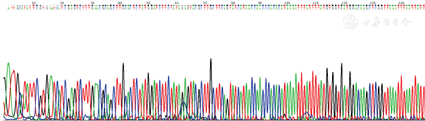
6种提取方法所得4种真菌(马尔尼菲青霉包括酵母相和霉菌相)DNA模板用真菌18S rDNA引物扩增后,PCR扩增产物5 μL经1.5%琼脂电泳,上述6种方法都可以扩增出阳性条带(图2),除白假丝酵母菌外其他3种真菌PCR阳性产物测序序列经BLAST比对后,18S rRNA同源性>99%,马尔尼菲青霉和小孢根霉测序图见图3和图4。
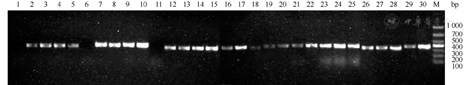
注:M.DNA标准带;1~5.加热裂解法提取各真菌DNA扩增产物;6~10.微波法提取各真菌DNA扩增产物;11~15.反复冻融法提取各真菌DNA扩增产物;16~20.试剂盒提取各真菌DNA扩增产物;21~25.溶壁酶法提取各真菌DNA扩增产物;26~30.蜗牛酶法提取各真菌DNA扩增产物。其中1、6、11、16、21、26为马尔尼菲青霉霉菌相;2、7、12、17、22、27为马尔尼菲青霉酵母相;3、8、13、18、23、28为白假丝酵母菌;4、9、14、19、24、29为新型隐球菌;5、10、15、20、25、30为小孢根霉
临床上确诊IFD主要通过真菌分离培养、表型鉴定,但这种方法耗时长,对专业技术要求高,结果判断的主观性较强,已逐渐不能满足临床诊断的需要。分子生物学技术的进步为IFD的诊断提供了新的思路,大大提高了检测的敏感性和特异性,且操作简便快速,目前有条件的临床实验室多采用PCR结合测序方法将真菌鉴定到种的水平,而PCR扩增首先要提取基因组DNA。然而,由于真菌细胞壁的主要成分为多糖,如几丁质、纤维素、葡聚糖等,酵母菌以葡聚糖为主,而丝状真菌细胞壁几丁质成分增加,比酵母菌结构更为坚固[10],因此,真菌基因组DNA提取需要增加破壁过程,故提取难度大于细菌及人体组织细胞DNA的提取。
当前真菌破壁方法主要包括机械法、酶法和化学法,其中机械法中液氮研磨法最为常用,但液氮研磨所需菌体较多,且研磨过程暴露在空气中容易污染,不适用于临床样本;化学法中大多采用十六烷基三甲基溴化铵(CTAB),氯化卞提取法等[11],由于提取过程添加了化学品,不仅污染环境,损害人体健康,而且这些方法操作繁琐,费时费力,限制了其推广应用[12]。为此,我们采用微波法、加热裂解法、反复冻融法、溶壁酶法、蜗牛酶法和试剂盒抽提法对不同类型的真菌,如温度双相菌(马尔尼菲青霉)、类酵母菌(白假丝酵母菌)、酵母菌(新型隐球菌)和霉菌(小孢根霉)分别提取基因组DNA,并从中筛选出适合临床实验室不同类型真菌基因组DNA快速提取的实用方法。
通过实验,我们发现6种方法均可成功提取基因组DNA,满足普通PCR扩增对模板DNA的要求。微波法提取真菌DNA浓度及产率最高,本实验用微波法提取小孢根霉、马尔尼菲青霉、新型隐球菌基因组DNA并进行PCR扩增、测序,均鉴定成功,且此法花费时间最短。但在操作过程中容易爆管,存在不确定性,并不适用于临床大批量操作。相比较而言,加热裂解法花费时间在15min左右,DNA产率虽低于微波法,但是操作简单,无需特殊设备,结果稳定,特别适用于临床的大规模快速检测。
从电泳结果看,蜗牛酶法与溶壁酶法提取DNA片段较完整,纯度也高,但是蜗牛酶法需对真菌进行过夜处理,花费时间最长,溶壁酶消化菌株时间短于蜗牛酶,但价格远高于蜗牛酶。两者最后都需用酚氯仿法提纯DNA,操作过程繁琐,需多次进行离心管转移,且这两种方法提取25℃培养下的马尔尼菲青霉时有时会一次纯化不成功,需多次纯化,不仅耗材而且DNA损失较多,结果受操作人员经验影响,不够稳定;这两种方法纯化DNA所用苯酚属致癌物质,气味难闻,对操作人员健康不利,还污染环境。Qiagen公司的DNeasy Plant mini kit,以酶法裂解真菌细胞壁,采用纯化柱纯化DNA,其所提DNA纯度高,与蜗牛酶法和溶壁酶法相比,纯化效果稳定,重复性好,花费时间少,且用试剂盒法提取时往往一次即能成功,适合本实验中所有真菌DNA提取,但提取过程中对真菌破壁不完全,或多次转移中DNA量损失过多,导致可能无法满足所需DNA浓度高的后续实验,且该法同样操作繁琐,费用较高。因此,蜗牛酶法、溶壁酶法及试剂盒法提取真菌基因组DNA并不适合临床实验室大样本快速分子鉴定真菌的需要。
本实验中,除了25℃培养下的霉菌相马尔尼菲青霉采用微波法、加热裂解法、反复冻融法提取基因组DNA经PCR扩增失败外,其他菌株用所有6种DNA提取方法均能提取到DNA并能成功进行PCR扩增,究其原因可能是马尔尼菲青霉在25℃培养下产生了水溶性红色素,而微波法、加热裂解法、反复冻融法所提DNA未纯化去除红色素,所以PCR扩增被抑制。因此,如果要提取马尔尼菲青霉基因组DNA,建议提取其酵母相的基因组DNA,从本实验结果看即使不存在色素抑制PCR反应问题,丝状真菌DNA提取还是难于酵母菌DNA提取。
目前PCR技术以其敏感、快速和特异性高等特点已被广泛应用于细菌、病毒的检测,在真菌检测方面也是一种非常有潜力的方法,与其他诊断指标联合应用,必能提高临床诊断效果[13]。临床实验室采用PCR结合测序分析鉴定真菌方便快捷,对罕见真菌的鉴定意义更大。在实际操作过程中,加快真菌基因组DNA提取速度、降低检测成本、简化操作流程可以缩短后续PCR检测及测序时间、加快真菌鉴定速度,对于临床早期诊断、早期治疗意义重大。本实验中采用6种方法提取真菌基因组DNA,发现微波法和加热裂解法提取真菌基因组DNA操作简单快速,能够满足临床实验室PCR检测真菌对模板的基本要求,其中微波法适合小样本快速检测,加热裂解法适用于大样本操作。

























