
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者(图1中Ⅲ2),女,28岁,因突发胸背部疼痛1 h入院。1 h前扫地时突发胸背部剧烈疼痛,不能忍受,伴大汗,无恶心、呕吐;无黑蒙、晕厥、意识障碍,为求诊治急来医院。查体:体温36.0 ℃,脉搏71次/min,呼吸频率18次/min,右臂血压158/86 mmHg(1 mmHg=0.133 kPa)、左臂血压125/72 mmHg。体型瘦高,神志清楚,精神差,急性病容,表情痛苦,端坐体位,推车入病房,查体不合作。长头瘦面,毛发分布无异常。颈软,气管居中,甲状腺不大。胸廓扁平,皮下脂肪薄。听诊:双肺呼吸音粗,双肺未闻及干湿性啰音,无胸膜摩擦音;心率71次/min,节律齐,各瓣膜区未闻及杂音。腹平软,无压痛及反跳痛,肝脾肋下未触及;MurpHy阴性。脊柱无侧弯,四肢细长,双下肢无水肿。心电图:窦性心律,ST、T改变。胸腹动脉CT血管造影(CTA):Ⅲ型主动脉夹层。此家族4代,共23人,现有10人,死亡3人,其中2人死亡原因不详,1人于田间劳动时突发胸痛猝死,考虑为主动脉夹层。其家系中发病者身材均瘦高,男性身高180 cm左右,女性170 cm以上,体型瘦高,四肢细长。
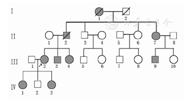
患者家族Ⅳ代均有发病,患者(Ⅲ2)的父亲(Ⅱ2)、1弟(Ⅲ3)及1妹(Ⅲ4)及其姑姑(Ⅱ7)、其姑姑的儿子(Ⅲ9)均被诊断为马凡综合征,患者的奶奶(Ⅰ1)为体型瘦高者,死因不详。患者(Ⅲ2)的另外1叔(Ⅱ3)1姑(Ⅱ6)与健康人结婚,其子女均无患病者。家系图谱分析:马凡综合征患者双亲一方患病,同一家族具有遗传并连续传递性,其Ⅱ代中男女均可发病,但健康同胞与健康人结婚的后代中无患病者,符合常染色体显性遗传病的家系特点。Ⅱ2家系发病率稍高,有遗传增强趋势。
马凡综合征(marfan's syndrome)又名蜘蛛指(趾)综合征,是在1896年由法国儿科医生Marfan首先报道,属于原因不明的以结缔组织为基本缺陷的遗传性疾病,为常染色体显性遗传。马凡综合征是一种常染色体显性遗传性疾病,主要累及全身结缔组织,造成心血管、骨骼、眼部病变。出现发育异常、脏器病变,甚至出现残疾或死亡。目前认为病因是原纤维素-1(fibrillin-1)编码基因突变所致[1],原纤维素-1是微纤维的主要组成成分,是广泛分散能执行多种功能的细胞外基质成分,由于基因突变改变了这种细胞外基质成分,出现弹性蛋白和胶原组织肽链之间的横向联合受损,酸性粘多糖沉积、唾液酸增多、透明质酸堆积、硫酸软骨素形成不良或过度破坏也有关。也有报道为营养不良、内分泌障碍及生化异常所致[2]。病变主要累及中胚叶的骨骼、心脏、肌肉、韧带和结缔组织。骨骼系统变化最常见,全身管状骨细长、手指和脚趾细长呈蜘蛛脚样。心血管方面表现为大动脉中层弹力纤维发育不全,造成大动脉扩张,形成大动脉动脉瘤或主动脉夹层。如大动脉扩张到一定程度,将引起大动脉大破裂死亡。本病常合并有二尖瓣关闭不全或脱垂、主动脉瓣关闭不全。眼部病变以晶状体全脱位或半脱位多见,主要是由于某种原因悬韧带断裂所致。
尽管致病基因的定位已经明确,但还没有特异的基因检测可用于马凡氏综合征的诊断,诊断以各个系统的临床表现和家族史为依据。其诊断标准[3]:①本病多见于青壮年;②有家族史,或家族中有猝死者;③眼部病变:晶状体向上脱位或半脱位;④心血管病变:有主动脉根部增宽,主动脉瓣关闭不全及二尖瓣脱垂等表现;⑤骨骼异常:表现为肢体细长、韧带松弛、脊柱侧弯以及漏斗胸等。
目前马凡氏综合征尚无有效治疗方法,日常生活中,患者应避免搬提重物、剧烈运动;平时注意眼睛保护,如戴护目镜。定期行心血管系统随访,以了解主动脉瘤变化,必要时行预防性的手术治疗,用人工血管行替代。眼科随访晶状体脱位、视网膜脱落等并发症情况。对孕期女性患者,更应加强心血管复查,有报道显示,主动脉直径<4 cm的女性怀孕分娩相对安全,>4 cm者,死亡风险增加。备孕时应咨询遗传学家和心血管专家相关事项,以降低发病风险。

























