
缺血性脑卒中是由于脑血流中断或显著减少而引发的一系列代谢和分子生物学改变,最终导致脑功能紊乱和形态改变。局部脑血流量(rCBF)和代谢的改变可通过放射性核素显像进行评估,尤其是SPECT和PET显像。SPECT和PET显像对脑血流和代谢阈值的评估加深了对脑功能和形态学改变的理解:PET在临床上脑卒中的缺血半暗带方面有很重要的应用,对制定治疗策略有很大影响。放射性受体显像可作为神经元不可逆损伤的早期标志物,并能预测梗死灶大小,这对大的("恶性")梗死灶是否进行有创治疗非常重要。在阻塞性动脉硬化疾病中,SPECT和PET显像可测定脑血流供应的储备量,这对制定干预治疗策略很重要;其同时还可检测病灶周围和对侧未受影响的脑组织,有助于了解脑功能网络受损而引起的临床症状。激活研究可以证明病变是否存在替代途径,并监测康复治疗的效果。放射性核素研究有助于发现神经炎性反应及其引发的进一步脑组织损伤。尽管放射性核素显像的临床应用存在一定的局限性,但该技术对脑血管疾病的研究仍具重要的意义,在对脑卒中的处理中有多方面的应用。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
脑血管疾病主要影响大脑的血液供应,而脑血流中的氧气和葡萄糖是神经组织维持神经功能的主要能量来源。因此,测量脑血流量(cerebral blood flow,CBF)是研究的重要一环。Kety和Schmidt[1]首次介绍了吸入惰性气体——氧化亚氮后分析多个动脉和静脉血液样本组织吸收率的定量方法来测量CBF。这种方法可以量化示踪剂在动脉输入和静脉流出间的差值,并提供每100 g脑组织中的平均血流量。随着放射性惰性气体注入到脑部供血动脉,示踪剂在皮质局部区域的积累和清除可通过外部放射性仪器进行检测,并通过分析清除曲线来确定该区域的局部CBF(regional CBF, rCBF)[2]。85Kr的β射线能量较低,需通过手术暴露皮质进行测量,因此很快被133Xe取代。133Xe的γ射线能量可穿过头颅而记录到清除曲线[3]。随后几年中,氙气法(颈内动脉或静脉注射氙气生理盐水或吸入氙气)成为测量rCBF的主要方法,测量脑灌注可使用多个探测器对大脑空间离散区域进行定量监测[4],或使用闪烁照相机[5]探测健康志愿者和各类脑疾病患者(包括脑血管疾病)[6]的二维血流图。SPECT[7]和PET[8]的发展提供了多种生理参数的三维脑显像图。目前这些技术在核医学中应用较广,该综述将总结SPECT和PET在脑卒中方面的应用。
神经组织的能量需求很高,因此必须保证脑组织有足够的血液供应。一个健康人的大脑包含约1 300亿个神经元(大脑皮质中为215亿个)[9],虽然其仅占人体质量的2%,却需要15%的心脏血液输出,约占全身总基础耗氧量的20%。正常生理条件下,大脑的葡萄糖氧化代谢几乎是脑内唯一的能量代谢[10]。神经元葡萄糖代谢主要是支持细胞营养和维持细胞功能。功能激活引起的血糖消耗增加(局部区域血流增多)主要集中在突触丰富的部位。总的来说,59%的总能量消耗用于突触传递,21%用于动作电位的传播,仅20%的消耗用于维持膜电位[11]。
脑血容量(cerebral blood volume,CBV)至少有3个组成部分:动脉血管、毛细血管和静脉。人类CBV中动脉约占30%,静脉和毛细管约占63%~70%。非扩散性显像剂用于CBV测量并不能获得脑组织灌注的确切值,但可评估局部血池的周转率。心脏输出血液的15%供应到大脑。正常全脑的CBV平均值约为50 ml·100 g-1·min-1;脑灰质的血流量较高,约为80 ml·100 g-1·min-1,白质平均为20 ml·100 g-1·min-1。脑氧代谢率(cerebral metabolic rate for oxygen,CMRO2)的平均值约为3.2 ml·100 g-1·min-1,灰质消耗约6 ml·100 g-1·min-1,白质消耗约2 ml·100 g-1·min-1。因此,正常的动静脉含氧量约差6.4 vol%容积,相当于一个正常血红蛋白浓度的人的颈内静脉血氧饱和度的65%~70%。葡萄糖是脑的主要能量底物,静息状态下脑内消耗的葡萄糖约占机体总耗糖量的25%。18F-脱氧葡萄糖(fluorodeoxyglucose, FDG) PET显像研究[12]发现,健康志愿者脑平均血糖消耗约为29~32 mmol·100 g-1·min-1,与Kety和Schmidt[1]测定的全脑代谢率相对应。大脑局部区域的代谢活动反映了脑的功能解剖区,最高值位于视觉皮质(45~50 mmol·100 g-1·min-1)和纹状体(42~46 mmol·100 g-1·min-1);大脑皮质其他区域及基底节(35~42 mmol·100 g-1·min-1)和颅后窝灰质(25~30 mmol·100 g-1·min-1)代谢活性均较低。最低的局部脑葡萄糖代谢率(regional cerebral metabolic rate of glucose,rCMRGlc)在白质区域为15~22 mmol·100 g-1·min-1[13]。大脑中,92%的ATP来自葡萄糖的氧化代谢,提供12 mmol ATP ·100 g-1·min-1。大脑中ATP和磷酸肌酸的储备只有8 mmol,少于1 min的储备。在无氧条件下,葡萄糖和糖原的无氧酵解可提供15 mmol的ATP。当脑组织氧分压下降至15~20 mmHg(1 mmHg=0.133 kPa)时,会出现意识丧失。当CBF完全中断时,10 s内即出现意识丧失。
为保持神经组织足够的血液、氧气和葡萄糖供应,脑循环中的稳态由以下几种机制严格调控:血流-代谢耦合(耦合到组织的功能需求)、CO2血管反应性(CO2分压)、氧分压(低氧血症引起的脑血管扩张)及自动调节机制(血压和脑灌注压)。
SPECT显像所用的放射性药物通常用于评估CBV、血流动力学和脑灌注。非扩散性显像剂(如131I标记的血清白蛋白)可检测由于脑血循环障碍导致的血管通过时间的变化,但无法获得组织灌注信息。因此,放射性显像剂需能够通过血-脑屏障并与rCBF的分布成比例,同时在脑局部区域具有足够的滞留时间以实现SPECT显像。对于大脑清除缓慢的显像剂,可通过微球模型来评估CBF。临床使用的放射性显像剂虽然不能满足所有要求(自由通过血-脑屏障、在血液中均匀分配、在大脑聚积而无再分配),但也能应用于临床[14]:123I-异丙基安非他明已商业化应用,注射111~222 MBq后其在脑内15~20 min达最大放射性,但由于光子能量较低,图像质量受限。99Tcm放射性药物具有很好的物理特性(140 keV,半衰期6 h),且核素发生器能在现场标记放射性药物六甲基丙胺肟(99Tcm-hexamethylpropyleneamine oxime,99Tcm-HMPAO)和双半胱乙酯(99Tcm-ethyl cysteinate dimer,99Tcm-ECD),两者在大脑中都能快速摄取,但存在轻度的首次通过清除效应,会造成rCBF偏低。99Tcm-HMPAO可在大脑中滞留,但在血液中清除较慢,因此灌注缺损可能较模糊;99Tcm-ECD在脑组织中摄取快且清除慢,而在血液中清除迅速,使其大脑的靶-本底比值高于99Tcm-HMPAO,图像质量有所提高[15,16]。99Tcm-HMPAO在体外不稳定,制备后须立即注射;而99Tcm-ECD在体外稳定可储存。此外,可通过吸入133Xe气体定量测量CBF,在低能量射线(81 keV)的衰变中可获得多个图像,但133Xe清除迅速,且需高灵敏度仪器进行探测[17]。
PET显像剂正电子与负电子结合发生湮灭辐射,产生2个方向相反、能量相等(511 keV)的γ光子,可用于符合计数。11C、15O和18F应用较广,其标记的各种化合物可用于生理和代谢变化的显像。
血浆和红细胞示踪剂可用于测量CBV,但血浆示踪剂68Ga-乙二胺四乙酸和11C-甲基化白蛋白由于血-脑屏障通透性的问题应用受限。11C或15O标记红细胞示踪剂CO已被广泛用于测量CBV,在健康受试者和患者中的测量值与其他方法的测量值一致性高[18]。11C标记的CO可一次大剂量吸入或连续快速2~3次吸入,15O标记的CO可单次或短暂吸入[19]或一次持续吸入(稳态15O模型)。
目前PET显像测量CBF的常用方法是基于显像剂15O-H2O在体内惰性自由扩散的原理[20],这种方法是通过单次"弹丸"式注射15O-H2O或15O-CO2后进行40 s的PET扫描来测量组织显像剂浓度。稳态15O法是使用15O标记的CO2或水持续给药。在扫描过程中,监测15O在动脉血中的放射性,并通过设定示踪剂的血-脑平衡分配系数来计算rCBF。
目前,有3种方法用PET测量CMRO2。最早的是15O连续吸入法[21],其中涉及到15O标记的CO2和O2的联合使用。该法通过吸入15O-CO2与15O2后分析图像的氧提取率比值来测量CBF,并通过15O-CO2扫描校正循环标记H2O。此外,为了获得准确结果,需用CBV扫描对血管内未摄取的15O进行衰减校正。第二种方法涉及到15O标记的H2O和O2联合应用来测量CBV、CBF和氧摄取率[22]。这种方法中,吸入O2通过像素运算方程来评估氧提取率像素值,这要求CBF像素与CBV像素相同。CBV通常是用单次吸入15O标记的CO来测量,CBF的测量则是标记H2O。此种方法迅速(共约40 min),相对于稳态法其对组织异质性不敏感。第三种方法是动态法,在单次或多次吸入或持续吸入氧分子后进行PET动态显像,可在单次研究中同时测量CBV、CBF、CMRO2和水的分布容积[23]。
18F-FDG的葡萄糖代谢研究是14C-FDG放射自显影技术的直接应用[24]。这种方法可以直接应用是由于18F-FDG与葡萄糖一样,在己糖激酶的作用下可转运到细胞内,18F-FDG被磷酸化为18F-6-磷酸脱氧葡萄糖。6-磷酸脱氧葡萄糖不能转化为6-磷酸果糖,也不能进一步降解为CO2和H2O,而在细胞内聚积。6-磷酸脱氧葡萄糖积累的动态变化可用计算rCMRGlc 3室模型的酶和转运常数表示[12]。
其他一些显像剂包括11C-氟马西尼、123I-碘西尼、11C-N-丁酮-2-乙基-1-(2-氯苯基)-N-甲基异喹啉-3-甲酰胺{N-butan-2-yl-1-(2-chlorophenyl)-N-methylisoquinoline-3-carboxamide,PK-11195}、18F-米索硝唑(misonidazol)也有特定方面的应用。
SPECT在脑血管疾病中的研究主要为脑灌注和rCBF显像,以及确认和扩展通过氙气技术获得二维CBF图像。由大或小血管病变引起的局部血液供应紊乱或栓塞引起的短暂性脑缺血发作或卒中的二维rCBF模式成像[25,26]可扩展为三维成像,并可与CT或MRI形态学图像配准。SPECT应用广泛,可评估脑组织中的血流灌注,但其常用的示踪剂(123I-异丙基安非他明、99Tcm-HMPAO、99Tcm-ECD)测得的rCBF较PET所测值可信度低。SPECT氙气法无需动脉血样即可定量测量rCBF,适用于在短时间内的重复研究,但该技术仅限于研究少数的脑血管疾病。只有PET可定量测量维持神经组织功能的有关生理变量及脑组织发生可逆的或不可逆损伤的相关生理变量,但PET测量应用较为局限,并需要动脉血样。尽管有这些限制,PET的研究使实验理论转移到临床应用成为可能,提高了对脑卒中病理生理学改变的认识,这是制定有效治疗策略的基础。
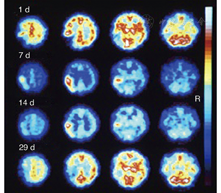
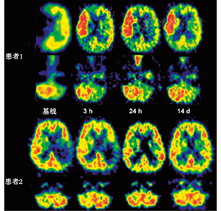
1.脑缺血病灶的检测。SPECT脑灌注显像及PET的血流和代谢研究[27,28]可探查缺血部位及缺血病灶的范围,这2种技术在检测梗死发生时间和梗死范围方面均比CT更为灵敏。在脑卒中发生后的第1个8小时,SPECT检测结果阳性率为90%,灵敏度为61%~74%,特异性为88%~98%[29]。早期PET研究显示,局部缺血部位存在血流和葡萄糖摄取差异,表明存在无氧糖酵解[30],测定氧摄取分数(oxygen extraction fraction, OEF)可使研究人员在梗死发生后的48 h内继续监测梗死的进展情况[31]。在此期间,增加灌注可观察到组织的不可逆损害,称为过度灌注[32];后期在梗死灶中心及周边区域可观察到相对充血(图1)。短暂性脑缺血发作可区别于缺血性卒中而发生在出现症状后的6 h内,SPECT在病侧的计数率为对侧的70%(脑卒中组织灌注值为对侧的35%~60%)[33];PET在病灶部位的CBF显著降低,但OEF增加[34]。
SPECT的检查结果与临床神经功能缺损及其症状的严重程度相关。早期严重缺血的SPECT检查结果较神经功能缺损评分严重,尤其是在考虑灌注缺损容积时,且其与CT扫描的梗死面积相对应[35]。总之,SPECT显像在卒中发病的5~8 h可根据脑灌注模式和耗氧量的变化对患者进行分类,评估预后并制定缺血治疗策略,虽然不如PET显像准确,但其对神经功能评分却有补充作用[36]。
2.缺血半暗带的识别。病理生理改变导致的功能障碍和组织不可逆损伤是急性缺血性脑卒中的治疗目标[37]。脑组织缺血阈值实验表明脑灌注降低分为2个等级:第1级为可逆性功能损伤的流量阈值(功能阈值);第2级的血流阈值较低,发生膜的不可逆破坏及形态损伤。血流灌注值在这两者之间的区域称为缺血半暗带[38],其特点是具有功能恢复的潜力且没有形态学损伤,提供充分的局部血流量后可以重建。当血流量低于该阈值时神经元功能立即受损,并随时间推移发生不可逆的形态损伤。这种相互作用的严重程度和缺血持续时间,在细胞发生不可逆损伤的同时,记录了皮质的神经元活动和rCBF[39]。这些结果对缺血半暗带概念进行了补充,即功能受损的神经元在缺血后的恢复潜力不仅取决于缺血期剩余血流量的水平,也与血流缺损的持续时间有关。
脑卒中早期,PET研究[40,41,42,43]发现脑缺血区域由不同的组织构成。测量时(通常在脑卒中发生后的几个小时)rCBF低于12 ml·100 g-1·min-1的组织或CMRO2低于65 mmol·100 g-1·min-1的局部区域在后期CT扫描中被发现是梗死区域。缺血区域残存的CMRO2是神经元相对完整的一个指标,预示该区域CBF将减少至12~22 ml·100 g-1·min-1。这种不完全的缺血灌注模式[41]是一个缺血半暗带,该区域的OEF增加(由正常值40%上升到80%)。PET研究将缺血区域分为3个部分:缺血核心区(12 ml·100 g-1·min-1),通常呈现出坏死;缺血半暗带区(12~22 ml·100 g-1·min-1),是仍然存活的组织,但可能发生梗死,也可能恢复功能;低灌注区(22 ml·100 g-1·min-1),没有受到血液供应不足的明显损害(图2)。缺血半暗带的进展及转化为梗死是一个动态过程,不可逆损伤的发生也是从缺血中心向周边进展。PET可对这个病理进展过程进行跟踪,并对动物模型和急性脑卒中患者的生理变量进行研究[44,45,46](图2)。
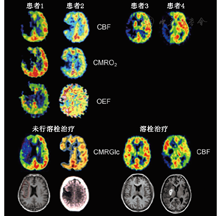
研究[47]发现在发作后的不同时间点,缺血半暗带累及范围取决于缺血发作的测量时间。在脑卒中后的第1个小时,测量脑血流量能够识别不同的组织,CT或MRI能识别梗死组织。把可能发生梗死的阈值设置为12 ml·100 g-1·min-1,则缺血半暗带的上限设定为18 ml·100 g-1·min-1,大部分梗死区域(70%)的灌注都低于12 ml·100 g-1·min-1;在缺血半暗带中(12~18 ml·100 g-1·min-1),一部分(18%)处在即将坏死的水平,一部分(12%)则处在一个较高灌注的水平。只有脑组织CBF高于12 ml·100 g-1·min-1时溶栓治疗才有意义。
SPECT灌注显像没有提供脑组织的代谢指标,故对残存组织是否进行再灌注治疗的评估比较困难。在缺血区域残存脑组织进行99Tcm-HMPAO再灌注时,脑组织相对小脑的摄取比值为0.55~0.75[48]。在卒中后的第1个小时,99Tcm-ECD的摄取率从45%变为52%,这些值对制定治疗策略可能有用[49]。通过Patlak曲线定量分析99Tcm-ECD SPECT摄取值的变化能够区分缺血的中心区域(20 ml·100 g-1·min-1)及周边区域(20~52 ml·100 g-1·min-1)[50]。99Tcm-HMPAO SPECT研究[51]发现,缺血区域残存脑组织中CBF高于35 ml·100 g-1·min-1的区域发生梗死的可能性较低,高于25 ml·100 g-1·min-1的区域通过早期再灌注可以恢复,但低于20 ml·100 g-1·min-1的区域则会发生不可逆损害。这些结果表明,SPECT脑灌注显像的结果与PET显像类似,可运用SPECT检查选择患者的治疗策略。
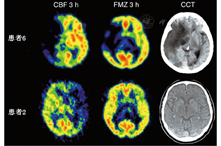
3.缺血半暗带的无创性显像。15O PET测量脑血流量与OEF需要动脉采血,而神经元完整性的测量无需动脉采血,可监测脑血管疾病发生不可逆损伤的不同时间点的生物标志物。氨基丁酸受体在大脑皮质表达丰富,能与中枢型苯二氮受体配体11C-氟马西尼结合。这些受体对缺血性损伤较敏感,可以识别早期的神经元损伤。11C-氟马西尼(图3)可以准确鉴定缺血性脑卒中早期的病理生理改变:在脑卒中发生后的第1个小时,55%的梗死区域有11C-氟马西尼摄取;21%的梗死区域血流量低于14 ml·100 g-1·min-1,而11C-氟马西尼的摄取高于临界值,表明该区域为缺血半暗带[44]。皮质的11C-氟马西尼摄取减低,可能是在缺血半暗带的周围或再灌注组织的缺血半暗区内有选择性的神经元缺失[52]。与SPECT显像剂对比,123I-碘西尼可在缺血性脑卒中的亚急性或慢性阶段对组织的不可逆损伤进行监测[53]。123I-碘西尼在缺血脑组织中的摄取与PET监测的CMRO2和OEF相关[54],且其在脑低灌注区的摄取能够滞留。123I-碘西尼SPECT显像中的梗死区域与MRI中的神经元缺失区域匹配[55],可用以判别哪些脑卒中患者梗死后从神经损伤中恢复会有较好或中度的预后。
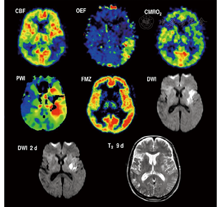
MR扩散和灌注成像能够区别梗死灶的中心和缺血半暗带。弥散加权成像(diffusion-weighted imaging,DWI)在早期显示的病灶为梗死灶核心,其邻近组织在灌注加权成像(perfusion-weighted imaging,PWI)上表现为低灌注。因此,PWI表现为低灌注而无明确界限的脑区(PWI-DWI不匹配区)定义为缺血半暗带[56],这种以显像不匹配区域来表示缺血半暗带的方法,DWI病灶几乎都为梗死组织,但PWI的数据采集并不完全准确,用于估算灌注的参数也是可变的且不完全正确。因此,脑灌注显示的病灶大小存在显著不同,且通常测量值偏大。达峰时间延迟4~6 s,能识别低灌注和正常灌注的组织,但会高估脑灌注减少但仍然存活组织的容积[57],因此在PWI-DWI上不匹配的容积(即缺血半暗带)与PET显像中显示的低灌注容积并不完全匹配(图4)。PWI参数与H215O PET血流量值的验证研究[58]可校正分类的准确性,但潜在可逆性、缺血最大容积、CBF和达峰值面积时间不能校正。
乏氧组织标志物可能识别缺血半暗带。18F标记的硝基咪唑类衍生物18F-氟硝基咪唑(fluoromisonidazole,FMISO)在动物实验后用于人类脑缺血研究[59],发现6例患者中有3例在梗死区周围有高摄取;在慢性期显像时发现高活性区消失,表明乏氧组织发生梗死或恢复。这种检测方法和监测病理进程在另一个研究[60]中也得到了证实。脑卒中发生后的6.25~42.5 h 18F-FMISO分布在梗死灶周围并扩展到正常组织。卒中发作后,18F-FMISO检测的乏氧组织量的改变可作为时间函数。最初受影响的组织容积、初始神经功能缺损的严重程度以及最初受累组织进展为梗死和神经系统疾病恶化的比例在脑卒中后的第1周内相关。18F-FMISO在这些区域的高摄取符合缺血半暗带的定义,故18F-FMISO高摄取的区域即缺血半暗带[61]。但18F-FMISO摄取的容积可能超出梗死区域,且梗死区域在早期可能没有显像剂摄取,表明18F-FMISO的摄取并不能准确区分可预防的发生梗死区域,因此不能准确定义缺血半暗带[62]。同时,显像剂的注入与显像所需的延迟(>2 h)进一步限制了18F-FMISO在急性缺血性脑卒中患者治疗决策制订中的价值。
缺血性脑卒中的治疗效果主要通过随机对照双盲临床试验证明,如静脉重组组织型纤溶酶原激活剂(recombinant tissue plasminogen activator,rtPA)的溶栓成功[63]。这样的对照试验需要收集大量的脑卒中患者,因此通常需很长的时间和大量的资金,在少数患者中用替代标记物可预测潜在的治疗效果,但替代标记物的应用又必须在有足够患者的对照试验中去验证。近年来,采用神经影像学识别在脑卒中可治疗修复的脑组织颇受关注,并作为脑卒中治疗效果的替代指标。
急性缺血性脑卒中的疗效监测也是建立在显像研究的基础上,缺血半暗带再灌注后可改善神经功能的损害(图5):患者接受rtPA治疗后的再灌注量与对照组相比显著增加[64]。在脑卒中发病后的3 h内,H215O PET CBF显像能够确定再灌注可修复组织的容积,并与梗死3周后MRI的梗死容积对比[65]。最初缺血的区域(即血流量低于阈值12 ml·100 g-1·min-1)再灌注后血灌注量接近正常水平,并预测在3周内临床症状得到明显改善。在一个12例的小样本研究中,灌注量低于脑卒中临界值的灰质,仅22.7%在溶栓治疗后会发生坏死,这意味着有很大一部分缺血组织再灌注治疗后仍可修复。另一个11例患者的PET研究[66]发现,只有当11C-氟马西尼在皮质摄取不减少或不低于白质平均摄取值的3.4倍时,缺血组织的再灌注才有效。这种神经元完整性的标志物可作为不适宜接受治疗的不可逆损伤组织的一个指标。
PET对筛查患者是否从治疗中获益的作用有限,SPECT脑灌注研究可在脑组织中任何区域简单、快速地检测和半定量分析脑缺血组织的CBF。99Tcm-HMPAO SPECT测定的CBF阈值可用于监测动脉溶栓的疗效[67]:脑组织中血流指数超过0.55的缺血组织,在症状发生6 h后仍可能修复;血流指数超过0.35的缺血组织,在症状发生5 h内仍可进行脑卒中治疗;而血流指数在0.35以下的脑缺血组织,尽管在早期就开始溶栓治疗但仍有出血风险。在静脉rtPA治疗中,99Tcm-ECD SPECT测得CBF为15%~53%,表明为不可逆损伤病灶;而CBF 45%~83%为可逆性局部缺血[68]。溶栓治疗后,99Tcm-HMPAO SPECT显像若局部脑血流低灌注表明预后较差,而治疗后为高灌注则预示症状改善,但不能预测脑出血的进展情况[69]。
有10%的大脑中动脉区域脑卒中患者会发展为恶性脑梗死。侵入治疗特别是去骨瓣减压术,可以降低这些患者的死亡率和发病率[70]。干预治疗是否有效以及干预治疗的最佳时间选择仍存争议,故需要更好地评估脑缺血不可逆损伤的方法。在34例患者中,超过50%的患者在大脑中动脉缺血区域的早期CT扫描上有变化,11C-氟马西尼PET显像可评估CBF和不可逆的神经组织损伤(图3)。随后,微透析探针和测量颅内压及组织氧分压的探针可分别进入同侧额叶[71]。在脑卒中发生后的24 h内,恶性病变患者(水肿形成与中线移位)与良性病变患者的病理改变相比,PET显像可见1个大的梗死灶核心(均值为144.5与62.2 cm3)以及体积较大的神经组织不可逆损伤病灶(均值为157.9与47.0 cm3)(图3)。在缺血病灶内CBF平均值明显降低,恶性病变的缺血半暗带体积明显小于良性病变组。在患者缺血病灶的恶性进程中,在症状发作后的22~72 h(平均为52 h)内脑灌注压下降至50~60 mmHg,随后,组织氧分压下降,谷氨酸增加,发生继发性缺血。谷氨酸、天冬氨酸、γ-氨基丁酸、甘油、乳酸-丙酮酸比值、次黄嘌呤、颅内压、脑灌注压、组织氧分压等监测值的变化达到最大水平。PET与实验研究相同,能够为恶性大脑中动脉梗死的颅内减压术提供早期干预治疗的时间窗;相反,侵入性监测并没有为恶性病灶的早期干预治疗提供一个足够早的时间窗。
小胶质细胞占大脑总细胞数的10%,在中枢神经系统受刺激时,其由静息态转变为激活态并具有吞噬功能。随后,相对分子质量为18×103的转运蛋白即过去称为外周型苯二氮类受体在激活的小胶质细胞线粒体中表达上调,因此可作为神经炎性病变的标志物。目前,一些放射性配体已开发应用于实验动物模型及各种中枢神经系统疾病的小胶质细胞活化的显像[72]。有研究[73]将11C-PK11195应用于高分辨率小动物PET显像,在脑缺血动物模型中发现,缺血后的第4天开始在病灶中心检测到高信号并持续增高至第7天,这个高信号与小胶质细胞和巨噬细胞有关。第7天,在梗死灶的周边可见一个不明显的高信号摄取,可能与葡萄糖代谢增加和小胶质细胞及巨噬细胞的积累有关;梗死灶周围的神经炎性反应可能会导致组织损伤的进一步扩展。
11C-PK11195在人类PET显像中发现,在脑缺血梗死1~2 d后,在梗死灶中心可见大量激活的小胶质细胞和巨噬细胞,随时间推移其从缺血中心向周边扩展[74,75]。有显像研究[72]发现,病变几天后,11C-PK11195在缺血病灶的边缘摄取增加,且在病灶的远处仍可见高摄取。在病灶同侧的丘脑和皮质下的白质特别是顺着锥体束通向病灶的区域可见显像剂的摄取也增加。在脑卒中后几周和几个月,这些区域将发生Wallerian变性[76]。外周炎性活动在一定程度上可发挥修复功能,而脑内激活小胶质细胞在梗死灶的中心和周边都与预后呈负相关。
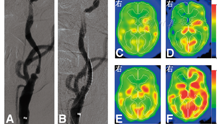
部分动脉血管性疾病引起的慢性灌注障碍患者处于非常危险的状态,故需血管外科手术。血管成像技术(如血管造影术)可以检测动脉闭塞或狭窄,并识别其侧支循环,但不能提供血液供应等代谢信息。只测量CBF存在明显不足,因为当脑血流灌注压降低时,其可代偿性舒张血管而保持正常的脑血流量。检测动脉阻塞的血流动力学和代谢改变的方法(尤其是颈动脉血流的改变),包括在基线测量CBF和应用血管扩张刺激剂后测量CBF(图6),因损害而增加的CBF表示减少的脑灌注压力;CBV和CBF的定量测量都在静息状态下进行,CBV/CBF比值增加表明血流动力学紊乱;测量OEF可作为局部自动调节功能是否正常的指标。在这些测量方法的基础上,学者[45,77]提出了血流动力学改变的3个阶段:0期,侧支循环完全弥补了动脉闭塞;Ⅰ期(血流动力学紊乱),其中CBV增加,但血管扩张刺激的CBF减小,OEF保持正常;Ⅱ期(血流动力学中断),CBF减少和OEF增加而保持CMRO2正常,以维持脑组织的代谢需求。OEF的异常改变可作为预测同侧后续脑卒中的指标之一[78]。在颈动脉支架置入术前和术后进行PET显像,可发现CBF、脑灌注压和CMRO2得到改善[79],SPECT显像血流动力学稳定(图6)[80],在双侧颈动脉疾病中也是如此[81]。代谢储备的降低可导致皮质神经元的丢失,这不能通过CT或MRI的形态学成像检测,但可通过123I-碘西尼SPECT[53]或11C-氟马西尼PET[82]发现。尽管部分患者存在这一阶段,但颅外-颅内搭桥手术并没有降低缺血性脑卒中患者在2年内复发的危险性,因为在围手术期脑卒中的发病率较高而抵消了长期受益[83]。
缺血性脑卒中病灶的早期PET研究[30]发现,其代谢和血流量减少的范围超过其受损组织形态学改变的范围,这与SPECT显像发现类似。最显著的表现是病灶对侧小脑的脑血流减少和代谢减低,称为交叉性小脑失联络[84],在脑卒中患者立即出现,在涉及小脑-皮质-脑桥途径而又成功地再灌注治疗的可逆性病变中持续存在(图5)[85]。交叉性小脑失联络是由一些神经元介导的效应,且因急性缺血而失活的部位已被证明位于小脑[86]。另外,在同侧皮质和基底神经节也存在脑血流的减少和代谢减低,但由于选择性的神经元缺失或血液供应不足也会影响到这些区域,故具体原因不明。然而,在非缺血性病变(如脑肿瘤和颅内血肿)中也会出现这种效应,因此相对于原发灶性质为何,其似乎与病变发生部位关系更为密切[84]。在皮质和皮质下病变中,顶叶和额叶梗死通常引起同侧基底节和对侧小脑的脑血流减少和代谢减低。该效应可通过皮质-脑桥-小脑途径损伤来解释。基底神经节的梗死可引起同侧大脑及对侧小脑失活。丘脑梗死主要影响病侧皮质,只有病变累及内囊时才会出现明显的小脑失活[87]。脑干和小脑梗死通常不引起前脑结构的不对称失活。交叉失联络不能通过常规的PWI参数检测到。临床症状可通过病灶远处的改变来解释,交叉失联络的程度与临床功能的缺损程度(脑卒中评分量表评估)与之后的功能恢复程度相关[88]。脑功能网络特定区域的失活和再激活在临床症状的出现和恢复中发挥着重要作用。
成人的脑梗死组织无法被替代或再生,因此,改善或恢复神经组织损伤只能通过重新激活功能受损但形态完整的部分,或寻找其在神经功能网络中的替代途径。在静息或脑功能网络激活的状态下,代偿机制可改变脑血流或代谢,通过放射性核素方法可监测其改变,如前述的氙气技术[89]和早期PET的应用[90]。神经影像学的研究包括在静息和激活状态下检测病灶部位和缺损的严重程度,比较其在激活状态下的功能改变。在脑卒中后早期和后期进行静息和激活状态下的纵向研究,可以揭示神经功能网络中受损的功能部分或完全恢复的代偿机制和替代途径[91]。
运动障碍的程度及其恢复潜力取决于病变部位、病灶在皮质区与纤维束的联系,以及脑深部灰质结构的累及(如基底节、丘脑和脑干)。改变脑代谢和血流量的方法及在给予血管扩张剂或运动后激活的方法有很多种,可通过其反映病变部位和严重程度,但这也依赖于给予刺激或运动方式的不同。
在大多数PET研究中,两侧大脑半球都有广泛的功能网络被激活。尽管在损伤和未损伤半球都可以观察到变化,但病侧运动皮质的活性是患者患侧手指功能从脑卒中后恢复运动的有利证据。相反,未受影响的手(如健康受试者)的运动主要由对侧大脑皮质的激活而控制。在运动皮质除了刺激的强度增加,其激活的范围也扩大,同时在病侧的运动前区和岛叶皮质也有激活。手臂恢复训练中可见在双侧顶叶、运动前区和对侧感觉运动皮质激活增加,提示双侧脑功能激活[92]。在皮质局灶性损伤后能够学习新的动作,表明存在更大的皮质区域,这主要取决于康复训练的强度。同时研究[93]表明,未受影响的半球可通过患侧手而抑制健侧手的自由运动,重复的经颅磁刺激可减少这种胼胝体抑制效应[94]。脑梗死灶的恢复也伴有患侧和健侧肢体的本体感觉系统的变化,反映出大脑刺激与恢复相关的本体感觉刺激存在分离。
脑卒中后失语的患者,右利手者的语言优势半球在左侧,左颞区是最常受损的部位,其损伤程度与失语症的严重程度相关。在脑卒中早期,rCMRGlc检测到语言相关脑区的功能性障碍是失语症的最终结果。此外,在梗死侧半球其他部位的代谢也与脑卒中后失语的结果相关[95]。偏侧大脑半球功能网络对预后有显著影响。当左侧大脑半球的语言中心受损时,尽管右侧大脑半球语言处理区代偿工作,但研究[96]结果发现成人这种方式的效率比语言相关功能网络修复的效率明显要低(图7)。在随访中,皮质下及额叶病变的患者基本得到改善,颞上回左缘实质区域的功能活性恢复;而颞叶病变的患者只有在语言恢复训练后才能好转,且颞上回左缘无法再激活。患者脑卒中后失语症的恢复只能通过在右侧额下回使用抑制性重复磁刺激来提高,这有利于左侧半球语言功能网络的恢复:PET观察到在失语症患者测试中有功能活性恢复提高的改变[97]。

由于放射性核素显像涉及复杂的流程环节,特别是生理学及分子变量的定量分析,其在脑卒中和闭塞性脑血管疾病的临床应用中受到一定限制。在急性脑血管病的临床应用中,要求像CT和MRI检查一样能直接、简单地获取图像,这些成像方式是脑卒中医疗护理的主要手段。但分子影像特别是PET显像,在缺血性脑损伤的病理生理学研究和发展新的治疗策略中一直都发挥着举足轻重的作用,通过无创性的方式获得病理生理改变的指标。
多模态显像将PET的生理和代谢信息与CT或MRI图像的解剖结构与病理改变相匹配。MRI是健康人和疾病患者脑形态学显像的最佳方法;而不同的MRI方法可用于评估生理和代谢参数的变化,如脑血管供应(增强MRI)、灌注成像(PWI)、脑水肿(DWI)、功能激活(功能MRI)及底物浓度的改变(磁共振波谱)。PET与MR融合成像已应用于实验研究,能同时在PET(皮摩尔)和高分辨率(微摩尔)MRI中评估代谢改变,这在临床上极具应用潜力[98]。
PET/MR能比较相同生理状态下PET和MRI灌注成像的测量值。其能区分梗死中心和缺血半暗带,并识别PWI-DWI不匹配区域。随着梗死时间的延长,可用于确定脑卒中后的最佳治疗时间窗。脑卒中的病变血管可通过磁共振血管造影(magnetic resonance angiography,MRA)检测,脑血流灌注或氧合血红蛋白和脱氧血红蛋白的变化(血氧水平-效应)与缺氧程度(15O)、乏氧(18F-FMISO)和代谢指标的变化(18F-FDG PET、乳酸磁共振波谱、胆碱、N-乙酰天冬氨酸),这些互补信息对制订治疗策略非常重要。高分辨率解剖图像信息由扩散纤维束成像补充,其与神经递质的激活或抑制和神经受体的活性有关,同时也与神经炎性反应相关。磁共振波谱和PET显像的联合应用,一方面可以检测葡萄糖代谢中的其他重要分子(如乳酸和丙酮酸),另一方面能够检测在急性脑缺血中的无氧糖酵解。
一些新的显像策略可能来源于PET/MR从基础研究到临床应用的转化过程,如:18F-低聚半乳糖-精氨酸-甘氨酸-天冬氨酸(Arg-Gly-Asp, RGD;18F-低聚半乳糖-RGD环肽生长因子受体)结合MR动态增强显像可能提供脑卒中后新生血管显像,从而为血运重建过程提供信息[99]。监测移植后的干细胞或祖细胞的位置和迁移,对发展各种神经系统疾病的细胞替代治疗策略非常重要。这些细胞可以被氧化铁颗粒标记,可通过MRI监测其在缺血部位是否存活及迁移的情况[100]。示踪细胞的MRI及验证细胞生物活性的PET显像联合应用可检测细胞生存能力及其是否整合到神经功能网络中的情况。从这些方面来看,放射性核素显像在脑缺血研究和脑卒中患者的临床治疗中仍有很好的前景。

























