
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
二肽基肽酶Ⅳ(DPP-4)抑制剂可抑制DPP-4活性,有效减少胰高血糖素样肽1 (GLP-1)的失活,以葡萄糖浓度依赖的方式促进胰岛素释放,从而降低血糖[1]。目前已在中国上市的DPP-4抑制剂有以下5种:西格列汀(sitagliptin )、维格列汀(vildagliptin )、沙格列汀(saxagliptin )、利格列汀(linagliptin)和阿格列汀(alogliptin )。本文将对上述5种DPP-4抑制剂的药化性质和药理活性之间的比较,及其差异对临床使用的影响作一综述。
根据DPP-4抑制剂的母核结构的不同,可将其分为拟肽类和非拟肽类两类。西格列汀、维格列汀和沙格列汀均为拟肽类抑制剂,是通过模拟GLP-1的N端二肽结构,然后经过结构修饰得到的,其中西格列汀的骨架为β-丙氨酸,维格列汀和沙格列汀的骨架为α-甘氨酸。利格列汀和阿格列汀为非拟肽类抑制剂,是通过高通量筛选再经过进一步修饰得到的,其中利格列汀骨架为含氮杂环黄嘌呤,阿格列汀骨架为嘧啶二酮。它们结构上的差别导致与DPP-4结合方式的不同。维格列汀和沙格列汀通过可逆的共价键与DPP-4结合[2],这种共价结合增强了与靶标的亲和性,这是共价抑制剂表现其高生物活性的根本原因。然而如果脱靶,共价抑制剂这种亲和性增强作用也会同样发生在脱靶靶点上,从而也带来增强的毒副作用。而西格列汀、利格列汀和阿格列汀通过非共价方式与DPP-4结合[2],可以避免共价抑制剂过度的亲和作用所导致的毒副作用。
除DPP-4以外,DPP家族还包括另外3个具有酶活性的成员,分别是DPP-8、DPP-9和FAP-α(成纤维细胞活化蛋白-α)[3]。DPP-4活性位点中包含Ser630的S1口袋在DPP-4、DPP-8以及DPP-9中均有存在,属于三者高度同源的结构。因维格列汀和沙格列汀中的氰基吡咯烷结构与Ser630可共价结合,导致二者对DPP-4/8/9都能发生抑制,故维格列汀和沙格列汀对DPP-4的特异性欠佳[4]。阿格列汀采用直接针对DPP-4活性位点结构的设计,使其能精确嵌入到DPP-4的活性部位,阿格列汀对DPP-4的选择性抑制是DPP-8/9或FAP-α的14 000倍之多,对DPP-4的特异性较好[5]。西格列汀的特异性不及阿格列汀,对DPP-4/DPP-8和DPP-4/DPP-9的选择性比值分别为大于2 660倍和大于5 550倍[5]。利格列汀对DPP-4选择性的抑制仅为FAP-α的89倍[6](表1)。
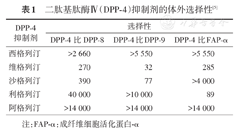
二肽基肽酶Ⅳ (DPP-4)抑制剂的体外选择性[5]
二肽基肽酶Ⅳ (DPP-4)抑制剂的体外选择性[5]
| DPP⁃4抑制剂 | 选择性 | ||
|---|---|---|---|
| DPP⁃4比DPP⁃8 | DPP⁃4比DPP⁃9 | DPP⁃4比FAP⁃α | |
| 西格列汀 | >2 660 | >5 550 | >5 550 |
| 维格列汀 | 270 | 32 | 285 |
| 沙格列汀 | 390 | 77 | >4 000 |
| 利格列汀 | 40 000 | >10 000 | 89 |
| 阿格列汀 | >14 000 | >14 000 | >14 000 |
注:FAP-α:成纤维细胞活化蛋白-α
临床前毒理学研究提示,DPP-8和DPP-9可能参与T淋巴细胞的激活与增殖,故抑制DPP-8/9可能与机体的免疫毒性相关,如在大鼠模型中出现了脱毛、血小板减少、网织红细胞减少、脾肿大甚至死亡的毒性反应[7]。Saisho和Itoh[8]报道了一个病例,由维格列汀引起的血管性水肿,换用阿格列汀1 d后该不良反应消失。因此DPP-4抑制剂的特异性和选择性可能会影响到该药物生物效应,对DPP-4具有较高选择性的DPP-4抑制剂,才有可能发挥最大药物疗效,避免因脱靶受体(DPP-4)而结合于其他非靶受体(FAP-α、DPP-8和DPP-9)所导致的潜在临床毒副作用。
DPP-4抑制剂经口服给药后迅速吸收,主要吸收部位在小肠,达峰时间在1~4 h之间。但5种药物的口服生物利用度差异较大:阿格列汀(100%)最高,西格列汀(71%~88%)、维格列汀(84%~85%)、沙格列汀(75%)属于中等,利格列汀(29.5%)最低[9]。因生物利用度反映了进入血循环的药量占给药量的比例,故生物利用度越大,表明进入血循环药量所占的比例越大,其能发挥的药效也相应越大。
除利格列汀与血浆蛋白结合的能力非常强(> 80%)外,其他DPP-4抑制剂与血浆蛋白结合的能力均比较微弱[10]。西格列汀、维格列汀、沙格列汀、利格列汀以及阿格列汀的表观分布容积(Vd)分别为198、71、151、368~918以及300 L,均大于全身含水量(42 L),提示该类药可在组织中广泛分布[11]。临床前研究显示,DPP-4抑制剂在肠、肾和肝脏浓度最高,也是DPP-4表达最高的组织。维格列汀、沙格列汀、利格列汀在脑内分布的水平非常低,提示它们可能不通过血脑屏障[5]。西格列汀、维格列汀、沙格列汀以及阿格列汀可能自由地透过胎盘屏障[5],由于在临床的数据较少,妊娠患者目前不推荐使用。
西格列汀、利格列汀、沙格列汀和阿格列汀的代谢机制与细胞色素氧化酶P450(CYP)有关,其中西格列汀、利格列汀和阿格列汀体内代谢较少,约80%的药物以原型形式排出体外。大约20%的西格列汀主要通过CYP3A4,其次是CYP2C8途径转化为6个代谢产物[12]。18%的利格列汀通过CYP3A4代谢为无活性的S-3-氢氧哌啶衍生物[13]。低于1%和大约5%的阿格列汀分别代谢为无活性的N-去甲基以及N-乙酰基阿格列汀[5]。而沙格列汀体内的代谢稳定性较差,大约50%的沙格列汀通过CYP3A4/5代谢成为具DPP-4选择抑制作用的5-羟基沙格列汀(活性约为沙格列汀的一半)[14]。肝功能受损会导致药物代谢变慢,可能会产生蓄积作用,故西格列汀和阿格列汀在重度肝功能不全患者中不推荐使用。沙格列汀在中度肝功能不全患者中需慎用,而在重度肝功能不全患者中则不推荐使用。维格列汀的代谢机制与CYP无关,55%的维格列汀在肝脏、肾脏以及其他组织中氰基水解为无活性的羧酸代谢物[15]。但临床发现维格列汀可能对肝功能有一定影响,所以禁用于肝功能不全的患者(表2)。

药物的半衰期反映药物在体内的作用时间及消除速度。西格列汀、利格列汀和阿格列汀的终末半衰期分别为8~14 h、120~184 h和21.1 h[9],推荐用药频率均为每天一次。利格列汀在体内为双相消除,消除半衰期可达120 h以上,但就目前临床文献未见利格列汀过量蓄积的报道。沙格列汀终末半衰期较短,仅为2.2~3.8 h[9]。因其通过共价键与DPP-4形成的可逆复合物,解离过程缓慢,其主要代谢物具有原型药物约50%的DPP-4抑制活性[14],也可以满足每天一次的给药频率。维格列汀也是一种与DPP-4共价结合的强效抑制剂,其终末半衰期仅有2.8 h,每天给药两次才可达到对DPP-4的有效抑制活性(抑制率≥80%)[17]。
西格利汀、沙格列汀、利格列汀和阿格列汀给药后24 h的DPP-4抑制率和最大抑制率分别为>80%和97%、~70%和80%、~70%和80%、~75%和90%[5],并且它们可将GLP-1的水平分别提高至安慰剂的2.0倍、1.5~2.0倍、2.0~3.0倍和3.0~4.0倍[9]。但是维格列汀推荐的最大剂量给药24 h后, DPP-4抑制率会降至35%[18]。所以,维格列汀与上述其他DPP-4抑制剂不同,需要每日服药2次来维持24 h疗效。
目前已有的5种DPP-4抑制剂既不是CYP酶的诱导剂也不是抑制剂,但西格列汀、沙格列汀、利格列汀以及阿格列汀均会不同程度地被CYP酶代谢。在与CYP酶相关的药物(如抗菌药克拉霉素、红霉素、甲硝唑,心血管药维拉帕米、辛伐他汀、华法林,降糖药二甲双胍、格列苯脲和吡格列酮,抗真菌药酮康唑、氟康唑,抗胃溃疡药物西米替丁,免疫抑制剂环孢霉素,糖皮质激素地塞米松,精神药物卡马西平、苯巴比妥)联用时可能会发生相互作用。但目前尚未发现西格利汀和阿格列汀具有临床意义的CYP相关的药物相互作用。沙格列汀与酮康唑(CYP3A4/5的强效抑制剂)联用时药-时曲线下面积提高了145%[19]。因此当沙格列汀与CYP3A4强效抑制剂联用时,需减量至每日2.5 mg[19]。而利格列汀不推荐与CYP3A4诱导剂合用。
西格列汀、维格列汀、沙格列汀以及利格列汀都是P糖蛋白的弱底物,可能会与同时服用的药物相互竞争P糖蛋白转运蛋白,导致同为上述转运蛋白的底物的药物消除速率发生改变,如抗病毒药物利巴韦林、利托那韦,心血管药物地高辛、阿托伐他汀,免疫抑制剂环孢素,抗菌药物红霉素、利福平等。目前尚未发现西格利汀、维格列汀和沙格列汀具有P糖蛋白相关的临床意义的药物相互作用。但是,研究提示200 mg西格列汀与0.25 mg地高辛联用10 d后,发现地高辛的药代动力学发生改变,药峰浓度提高了18%,虽然不推荐调整地高辛的剂量,但是二者联用时需紧密监测。5 mg利格列汀与600 mg利福平联用时,与未联用相比,利格列汀的24 h内药-时曲线下面积下降了40%,血药峰浓度下降了44%。因此利格列汀不建议与P糖蛋白强效诱导剂联用[19]。
DPP-4抑制剂虽然具有相同的治疗糖尿病的靶点,但是各自的药化性质和药理活性还是有所差异,本文希望通过分析药物的特点,从而在一定程度上帮助临床医师选择合适的治疗药物。临床前的数据显示阿格列汀的特异选择性最高,能较好地避免脱靶导致的潜在的临床副作用。除维格列汀需要每日服用2次外,大部分DPP-4抑制剂每天服用一次即可达到良好的DPP-4抑制作用,并可显著提高内源性GLP-1的活性。维格列汀在肝功能不全的患者中不推荐使用,在重度肝功能不全的患者中,西格列汀、沙格列汀和阿格列汀不推荐使用。中/重度肾功能不全患者维格列汀不推荐使用,西格列汀、沙格列汀和阿格列汀需酌情减量,而利格列汀则不受影响。沙格列汀对DPP-4的抑制作用受CYP酶抑制剂或诱导剂的影响较大,联合使用时应酌情注意调整剂量,而利格列汀不建议与CYP3A4或P糖蛋白强效诱导剂联用。
综上所述,DPP-4抑制剂作为一类新型降糖药物,大量研究已证实它能有效地抑制DPP-4,延长GLP-1的降糖疗效。然而不同DPP-4抑制剂在临床应用的差异,还有待更多的临床数据及真实世界的临床治疗的观察。
利益冲突声明 作者在文章撰写过程中接受了赛诺菲(中国)提供的编辑/撰写支持。但是,作者对所有内容及编辑决策负完全责任,并未就撰写文章本身接受任何其他形式的酬劳

























