
回顾1例以癫痫发作起病的线粒体tRNA Leu(UUR)A3243G基因突变糖尿病。该病以母系遗传、血糖升高、多伴有听力障碍为特征,可合并全身多系统疾病,具有遗传倾向。临床如未及时诊治,可致家族聚集发病。临床医生应提高对此病的认识,早期识别,做好优生监督。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
线粒体基因突变糖尿病,又称母系遗传糖尿病伴耳聋(maternally inherited diabetes and deafness,MIDD),是由呼吸链缺乏、胰岛β细胞功能缺陷所致的一组以母系遗传、血糖升高、多伴有听力障碍为特征的糖尿病,以线粒体亮氨酸转运RNA[tRNALeu(UUR)]上的线粒体核苷酸序位3243上的A➝G(A3243G)突变最为多见,除此之外,nt3316G➝A、nt3394T➝C、nt16198T➝C等也是常见的突变型。世界卫生组织及中国指南将其归类为胰岛β细胞功能缺陷所致的单基因突变糖尿病,国内突变率为0.4%~1.8%,国外为0.5%~2.8%[1, 2, 3]。除MIDD外,该突变还可出现在线粒体脑肌病伴高乳酸血症和卒中样发作(mitochondrial encephalopathy lactic acidosis stroke-like episodes,MELAS)、肌阵挛癫痫伴破碎红纤维综合征、进行性眼外肌麻痹、肥厚性心肌病等多种疾病[4]。临床如未及时诊治,可致家族聚集发病。笔者结合1例tRNALeu(UUR)A3243G突变型MIDD患者的临床资料,对该疾病的研究进展、临床表现、诊断及治疗进行分析。
患者 男,汉族,17岁。因“抽搐2周”于2017年7月20日就诊于我院。患者2周前无明显诱因出现发作性意识丧失伴肢体抽搐,伴精神异常,发作时双眼斜视、双手握拳、四肢抽搐、意识丧失,持续数分钟,完善颅脑磁共振检查示右颞顶叶皮层区多发性脑梗死灶可能性大,小脑和脊髓萎缩。给予奥卡西平300 mg,每日两次,未再发作。2年后患者出现消瘦,体重减轻10 kg,无烦渴、多饮、多尿、胸痛、呼吸困难等。出生后运动发育、智力及生理发育与同龄儿无异,即将完成高中学业,成绩一般,性格易怒。家族史:母亲、外婆患糖尿病,均30余岁起病,否认家族中其他成员患此病及其他遗传相关性疾病。体格检查:身高168 cm,体重55 kg,体质指数19.49 kg/m2,体重最高65 kg,血压正常。神清,反应稍迟钝,记忆力、计算力较差,理解力、定向力一般,双侧眼睑无下垂,眼球活动自如,眼震(+/-)。双侧瞳孔等大等圆,对光反射灵敏。双侧鼻唇沟无变浅,伸舌居中。视力:左眼0.8,右眼1.0;颈软,无抵抗。心肺腹查体未见明显异常。四肢肌力5级,肌张力正常,腱反射(+),脑膜刺激征、病理征未引出,指鼻试验、轮替试验欠稳准,深浅感觉无异常。辅助检查:空腹血糖11.87 mmol/L,糖化血红蛋白13%,C肽3.44 ng/ml,胰岛素14.08 mIU/L,肌酸激酶205 U/L,肌酸激酶同工酶75 U/L。动脉血气:pH 7.37。乳酸1.7 mmol/L。尿常规:尿糖(3+),酮体(+),蛋白(+/-);尿微量白蛋白/肌酐39.38 mg/g。超声心动图:左心室舒张末期内径5.2 cm,左心室后壁舒张期厚度1.2 cm,左心室射血分数50%,左心室扩大,左心室心肌肥厚,二尖瓣反流,左心室舒张与收缩功能减低。眼科及听力检查未见明显异常。基因检测方法:采集患者外周血2 ml,-20 ℃低温保存备用。由青岛金域医学检验所利用QIAamp DNA试剂盒提取外周血的基因组DNA,经纯化、扩增、测序后筛选出3243位点所在的mt.3150~3403为可疑突变片段,设计引物进行PCR扩增后利用Sanger法基因测序,可见mt.3243A>G杂合突变,变异频率约为56%(图1)。
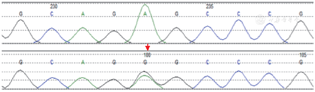
此例患者以癫痫发作起病,入院后超声心动图提示心肌肥厚,2年后出现血糖升高,有明确母系家族史,结合常规实验室和线粒体基因检查,确诊为MIDD。就诊后嘱患者低碳水化合物饮食,给予阿卡波糖100 mg每日3次、沙格列汀5 mg每日1次治疗至今。门诊随访:空腹血糖(1-2-3-6个月)5.40-7.30-6.14-6.09 mmol/L,糖化血红蛋白(1-2-3-6个月)9.5%-5.1%-5.8%-6.3%,C肽(9个月)2.07 ng/ml,胰岛素(9个月)7.18 mIU/L。
线粒体tRNALeu(UUR)A3243G基因突变型发现于1992年,最初由Ballinger等[5]在一母系遗传的糖尿病家系中获得。同年,van den Ouweland团队利用分子生物学技术发现此为tRNALeu(UUR)A3243G突变,该突变最早证明与MELAS相关[6]。随后多国学者在非胰岛素依赖型糖尿病患者中发现此类突变,我国首例由上海市糖尿病研究所行基因诊断初筛时获得[7]。
MIDD的病理生理变化主要发生在线粒体内。线粒体的氧化磷酸化是细胞供能的中心环节,包括胰岛β细胞、脑细胞、小血管平滑肌以及内耳血管边缘细胞在内的各种器官组织均需此过程提供三磷酸腺苷(adenosine triphophate,ATP)。以胰岛β细胞为例,线粒体DNA的环状双螺旋结构包含介导氧化磷酸化的基因。正常情况下,血浆中的葡萄糖通过氧化磷酸化产生ATP,并转运至胰岛β细胞内,使胞内ATP/二磷酸腺苷水平升高,依赖于ATP的K+通道关闭,膜电位极化,继而Ca2+通道开放,Ca2+进入细胞内,使细胞内的Ca2+浓度增加,细胞分泌胰岛素。而在此类疾病中,A3243G突变位于tRNALeu(UUR)的双氢嘧啶环上,该位点前后的13个碱基是线粒体转录因子的结合位点,此突变不仅改变tRNALeu(UUR)的双氢嘧啶环,影响tRNALeu(UUR)的合成,同时也使线粒体末端转录异常,使氧化磷酸化基因的翻译过程出现障碍,ATP合成减少,最终影响葡萄糖介导的胰岛素分泌;另外,突变致高毒性OH-增多,通过氧化应激引起DNA破坏,也可能是致病的重要原因[8]。以上过程发生在胰腺,导致血糖升高,发生在其他器官组织,则会致癫痫发作、卒中样表现,甚至听力受损,最终发生与基因突变相关的多种综合征。
MIDD的临床表现有:(1)母系遗传;(2)发病早,平均发病年龄在30岁以下;(3)绝大多数为非肥胖型;(4)谷氨酸脱羧酶抗体阴性;(5)感音神经性耳聋,以高频为主,听力呈进行性下降;(6)胰岛素分泌功能进行性衰退,胰岛素抵抗不明显;(7)合并其他系统疾病,可累及神经肌肉系统(如癫痫、卒中样发作等)、心肌(如心肌病、传导阻滞)、视网膜、肾脏等,发病趋向年轻化[9]。本研究中病例符合以上除耳聋外的大部分临床表现,临床诊断MIDD较容易。但由于患者以癫痫发作起病,虽有抽搐症状,但查乳酸水平、颅脑磁共振成像等均与MELAS不符,首诊为神经内科,易被误诊为脑炎、单纯癫痫发作、脑梗死等延误治疗;其次,患者起病早,在未完善基因的情况下,需与其他基因遗传病相鉴别。
MIDD的个体化管理仍在探索,现有的治疗包括:(1)饮食:该例患者目前选择生酮饮食,随访半年内未出现MELAS相关症状,复查胰岛功能在正常范围内有所下降,可能提示胰岛细胞获得休息,不再保持高分泌状态,也可能是患者的胰岛功能在进行性衰减,仍需进一步随访观察。有研究指出,生酮培养基可选择性干扰呼吸链功能,提高线粒体蛋白质合成能力,因此,生酮饮食具有较好的实验价值;但也有专家提倡适当放宽饮食控制。(2)运动:因本病致血清乳酸水平升高,患者不宜剧烈运动,提倡有氧运动为主。(3)药物:①磺脲类药物可用于早期胰岛功能尚可的患者,随着胰岛功能进行性衰退,应尽早应用胰岛素治疗;避免使用二甲双胍,以免发生乳酸酸中毒。②有少数应用泛醌、泛癸利酮改善胰岛β细胞功能的案例。③环孢素可预防线粒体DNA所致心脏疾病。④应用辅酶Q10、线粒体辅助因子(肉碱,维生素B、C、K等)、肌酸改善呼吸链功能。在该患者的诊疗过程中,根据胰岛功能,选择α-糖苷酶抑制剂、二肽基肽酶4抑制剂降糖,随访时血糖控制良好;患者体型消瘦,继续监测血糖及胰岛功能,必要时应用磺脲类或胰岛素,促泌或替代者治疗的同时,还可达到增重目的。(4)基因疗法:是治疗线粒体疾病的根本方法,某些基因层面的干预手段也取得一定成效[10, 11]。另外,妊娠女性有早产和胎盘植入的风险,应在妊娠晚期严密监测,避免使用硫酸镁加重肌肉损伤[11]。
自线粒体tRNALeu(UUR)A3243G基因突变糖尿病被发现后的近30年间,人们对这一疾病的病因、病理生理、诊断及治疗的认识更加深刻,由于tRNALeu(UUR)上的3243位点发生A➝G突变,导致线粒体功能异常,表现于不同靶器官而形成综合征。该患者以癫痫发作起病,却无典型的MELAS相关证据支持,在之后随访中,逐渐出现血糖异常、心肌肥厚等表现,才将研究者的关注点集中于MIDD。治疗方面以生酮饮食联合α-糖苷酶抑制剂、二肽基肽酶4抑制剂为主,目前的随访结果暂不能说明患者的胰岛功能变化,仍需观察。作为一种基因突变类疾病,针对基因治疗方面的研究需进一步开展,以期达到根治疾病或改善预后的目的。通过回顾此病例,希望增加临床医生对该疾病的认识,在更好的根治方法问世前,做好早期诊断,优生监督。
所有作者均声明不存在利益冲突

























