
本文为回顾性研究,经南京鼓楼医院伦理委员会批准,免除受试者知情同意,批准文号:2022-CR005-01。
本刊刊出的所有论文不代表本刊编委会的观点,除非特别声明
本文为回顾性研究,经南京鼓楼医院伦理委员会批准,免除受试者知情同意,批准文号:2022-CR005-01。
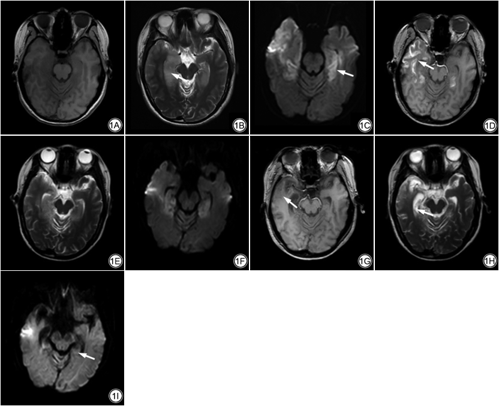
患者女,23岁,2021年6月16日无明显诱因下头晕不适,伴阵发性恶心呕吐,外院就诊示白细胞升高,予以抗炎治疗,症状稍好转。一日前突发言语混乱,记忆丧失,行为紊乱,伴发热,热峰38.8 ℃,遂至我院就诊,急查肝功能示钠128.9 mmol/L、氯91.6 mmol/L、尿素氮6.25 mmol/L、葡萄糖7.4 mmol/L、谷草转氨酶61 U/L、总胆红素25.4 μmol/L,脑电图示中度异常脑电图,额、中央区θ频段功率明显增强,6月18日以“精神异常原因待查”收住我院。6月21日头颅MRI示两侧颞叶内侧、右侧颞叶外侧及颞极、右侧岛叶片状等T1长T2信号,液体衰减反转恢复(fluid attenuated inversion recovery, FLAIR)呈高信号,扩散加权成像(diffusion-weighted imaging, DWI)示扩散受限,双侧海马肿胀提示自身免疫性脑炎或病毒性脑炎(图1A~1C)。同日查脑脊液N-甲基-D-天冬氨酸受体(N-methyl-D-aspartate receptor, NMDAR)抗体IgG 1∶1,血清NMDAR抗体IgG 1∶100,确诊为抗NMDAR脑炎。7月7日复查MRI示原脑实质内水肿病灶范围减小,并出现脑萎缩及病灶区沿脑回分布的线状短T1信号,其余序列信号较前相仿,考虑为新发脑皮质层状坏死(图1D~1F)。自6月21日确诊,患者采用丙种球蛋白0.4 g/kg•d联合甲强龙1 g冲击治疗(激素每5 d减半),7月11日查脑脊液NMDAR抗体IgG 1∶1,血清NMDAR抗体IgG 1∶10,7月12日出院前临时使用一次丙球抗体封闭血液中尚存抗体。8月18日复查MRI示上述水肿病灶进一步减小,脑萎缩加重,沿脑回分布的短T1信号消退,DWI扩散受限消退(图1G~1I)。
讨论 抗NMDAR脑炎是2007年[1]新明确的一种自身免疫性脑炎,约占自身免疫性脑炎的80%[2],青年女性好发。本病发病机制尚未明确,可能诱因为肿瘤、病毒等使机体暴露自身神经元抗原,机体通过体液免疫产生抗体,攻击神经元表面的NMDAR-GluN1亚基从而引起的免疫介导性疾病[3]。抗NMDAR脑炎可有单个或多个病灶,因此本病临床症状多样,多表现为急性或亚急性认知和意识的改变、癫痫、神经精神症状等,70%患者发病前可有前驱感染症状(发热、腹泻等)[4]。主要诊断依据为脑脊液及血清抗NMDAR抗体检测。患者的头颅MRI不具有明显特异性,通常表现为大脑皮层、脑干、小脑、基底节等区域T2WI或T2-FLAIR高信号,易累及颞叶内侧及额叶皮层[5]。病变脑区分布与病毒性脑炎有重合,本例患者存在发热,这为鉴别诊断带来困难,因此该类患者应尽快完善脑脊液检查。据报道,约1/3抗NMDAR脑炎患者会出现弥漫性脑萎缩,其发生的机制尚不明确,但这并不意味着长期预后差[6]。本例患者后续MRI检查发现脑萎缩,且在随访过程中出现进展,但并没有出现相应症状。
脑皮质层状坏死(cortical laminar necrosis, CLN)是一种特殊类型的皮质坏死,是多种原因引起的中枢神经系统氧和(或)糖的摄取障碍及脑能量代谢异常[7]。目前CLN发病机制尚不十分明确,有“血管学说”和“异感学说”两种解释:(1)“血管学说”认为大脑皮质的毛细血管按层状分布,而毛细血管的形态或功能异常都会影响脑皮质的血氧供给,从而引起不同程度的神经元损伤;(2)“异感学说”认为不同灰质结构及不同类型神经元的化学结构、受体和神经递质等对缺氧、兴奋性氨基酸等的耐受反应不同,其中第3层对血氧耐受最敏感,即皮质第3层最易受缺血缺氧、能量供应障碍等影响而发生CLN[8]。在MRI上,CLN表现为沿皮层分布的线状短T1信号,这是由坏死物中的变性蛋白与水结合后形成的高浓度蛋白溶液以及大量沉积的含脂巨噬细胞和坏死组织细胞成分所致[9]。导致CLN的病因很多,其中国内报告以脑梗死多见,也有肾上腺危象、渗透性脱髓鞘综合症、氰化物中毒等致使CLN的个案报道。抗NMDAR脑炎所致的CLN极为罕见,目前国内外文献尚无类似病例报道。推测本病例是由于自身免疫反应介导了神经元损伤,产生了类似缺氧的神经元损害,另外也可能与起病初期电解质代谢紊乱及肝功能异常导致的神经细胞能量代谢异常相关。
CLN的MRI表现具有特征性时间变化[10]:沿大脑皮质分布的脑回样T1WI高信号在病变发生后2周左右出现,此时易误诊为脑实质少许出血或蛛网膜下腔出血;T1WI高信号2周后逐渐显著,1~2个月时达到峰值,随后开始消退,最多可持续2年。本例起病2周左右复查MRI发现病灶区T1WI脑回样高信号,2个月后复查时消失,基本符合CLN的时间变化特征。
通过本病例的分析,我们不仅掌握了抗NMDAR脑炎的影像学特征,更重要的是扩展了对CLN的认识,避免了罕见的继发性表现对原发疾病诊断的干扰,使临床治疗少走弯路。
National Natural Science Foundation of China (No. 81971596).
全体作者均声明无利益冲突。

























