探讨VPS13B基因突变所致Cohen综合征患儿的临床特征及诊断特点,并进行相关文献复习,为Cohen综合征遗传咨询和诊断提供参考。
选择2019年1月,于中山大学孙逸仙纪念医院儿科确诊为Cohen综合征的2例患儿(患儿1、2)为研究对象。采用回顾性分析方法,收集2例患儿的临床病例资料,并对其病史采集、相关检查结果进行分析。以"Cohen综合征""Cohen syndrome"为关键词,检索在线人类孟德尔遗传数据库(OMIM)、PubMed数据库、中国生物医学文献服务系统(SinoMed)、中国知网(CNKI)、万方数据知识服务平台、维普中文科技期刊数据库中Cohen综合征相关文献,检索时间设定为1973年1月1日至2019年1月1日。本研究符合2013年修订的《世界医学协会赫尔辛基宣言》要求,并且征得受试儿家属(父亲、母亲及表叔)知情同意。
①患儿1病史采集:男性,4岁3个月,主要临床表现为精神运动发育落后、小头畸形和身材矮小。相关检查结果:血常规检查结果提示中性粒细胞计数减少;听性脑干反应(ABR)检查提示左侧听觉传导通路损伤,双侧听觉反应阈值可疑范围;双眼视觉诱发电位(VEP)检查未见异常;脑电图正常;头颅MRI平扫提示双侧额部脑外间隙稍增宽;全脊柱正侧位X射线摄片提示右侧轻度髋发育不良伴右髋关节半脱位可能;腰椎轻度左侧弯,疑为双髋关节不对称所致;心电图、心脏彩色多普勒超声检查和染色体核型分析均未见异常。②患儿2病史采集结果:患儿1胞弟,男性,1岁4个月,亦表现为精神运动发育落后,小头畸形和身材矮小。相关检查结果:双耳ABR正常;VEP检查提示右眼F-VEP P2波潜伏期稍延迟,振幅正常;脑电图正常;头颅MRI平扫未见异常;全脊柱正侧位X射线摄片、心电图、心脏彩色多普勒超声检查和染色体核型分析均未见异常。③患儿1、2遗传性疾病大家系全外显子组检测提示,2例患儿均携带VPS13B基因2个杂合病理性突变,即VPS13B[8q22 NM_017890.4 Intron50 c.9259-1G>C][8q22 NM_017890.4 Exon57 c.11104_11105de1]基因突变,这2个基因突变位点目前均尚未见文献报道。
Cohen综合征为一种罕见常染色体隐性遗传性疾病。临床上对于特殊面容、生长发育落后、高度近视、弱视和色素性视网膜病变伴中性粒细胞减少症的患儿,应考虑到Cohen综合征的可能性,并进行基因检测,以及时确诊Cohen综合征。


版权所有,未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别申明,本刊刊出的所有文章不代表中华医学会和本刊编辑委员会的观点。
本刊为电子杂志,以光盘形式出版。本册应读者需求按需印刷,随光盘免费赠阅。光盘如有质量问题,请向编辑部调换。
Cohen综合征是一种极为罕见的遗传性疾病,又被称为"脑-肥胖-眼-骨骼"综合征,于1973年由Cohen等首次报道。迄今为止,全球仅有少量Cohen综合征病例被报道[1,2,3,4,5,6,7],国内目前仅见2例该病患儿被报道[8,9]。该病临床表现复杂,诊断困难,目前尚无有效治疗方法,也无相关康复治疗研究报道。本研究对2019年1月,于中山大学孙逸仙纪念医院儿科确诊的2例Cohen综合征患儿进行回顾性分析,包括其临床表型、实验室检查及基因检测结果等,并进行相关文献复习,旨在提高临床医师对Cohen综合征的认识和重视,从而提高对该病的诊治水平。现将研究结果报道如下。
选择2019年1月,于中山大学孙逸仙纪念医院儿科确诊为Cohen综合征的2例患儿(患儿1、2)为研究对象,其年龄分别为4岁3个月和1岁4个月,为胞兄弟。本研究遵循的程序符合2013年修订的《世界医学协会赫尔辛基宣言》要求,并且征得受试儿家属(父亲、母亲及表叔)知情同意。
对于以下8项临床表现中,具有6项及以上者,即可诊断为Cohen综合征。①发育迟缓;②小头畸形;③具有头发、眉毛、睫毛浓密,睑裂下斜,凸出、喇叭样鼻子,人中短、上翘等典型的Cohen综合征面部特征;④四肢纤细、躯干性肥胖;⑤过度的社交行为;⑥关节过度伸展;⑦高度近视和(或)视网膜营养不良;⑧中性粒细胞减少[4]。若检测到VPS13B基因病理性突变,则可直接诊断为Cohen综合征。
收集2例患儿及其父母外周静脉血各2 mL,置于含乙二胺四乙酸二钠抗凝剂的采血管中,于4 ℃条件下保存,并送至广州金域医学检验中心进行基因检测。通过高通量测序技术,对基因全外显子区进行直接测序,并与参考序列(NM_017890.4)进行比较。此外,收集患儿表叔外周静脉血2 mL,置于含乙二胺四乙酸二钠抗凝剂的采血管中,于4 ℃条件下保存,并送至广州达安临床检验中心,对其VPS13B基因c.9259-1G>C位点及c.11104_11105de1位点进行验证。
以"Cohen综合征""Cohen syndrome"为关键词,检索在线人类孟德尔遗传数据库(Online Mendelian Inheritance in Man,OMIM)(https://omim.org)和PubMed数据库,中国生物医学文献服务系统(SinoMed),中国知网(China National Knowledge Infrastructure,CNKI),万方数据知识服务平台,以及维普中文科技期刊数据库中,Cohen综合征相关文献。检索时间设定为1973年1月1日至2019年1月1日。
患儿1(先证者),男性,4岁3个月,因"精神运动发育迟缓4+年",2019年1月3日于中山大学孙逸仙纪念医院儿科门诊就诊。病史采集结果:患儿系G1P1,晚孕期出现胎动减少,足月儿,经阴道自然分娩娩出,出生体重为2 800 g,身长为43.5 cm。该例患儿出生时因"羊水Ⅲ度浑浊、新生儿肺炎",于当地医院新生儿科接受住院治疗的检查结果:头颅MRI提示双侧苍白球对称性稍短T1信号,新生儿胆红素脑病改变待排查;心脏彩色多普勒超声结果显示房间隔缺损(左向右分流),动脉导管未闭(左向右分流)。患儿出院后于当地医院接受康复治疗。该例患儿分别于2岁、2岁3个月、2岁6个月、3岁1个月和4岁时,罹患肺炎。生后4+个月龄时抬头稳,8+个月龄时可独坐,约1岁时可爬行,约2岁6个月时可独站和能叫"爸爸"和"妈妈",但是该例患儿于本院就诊时仍然不能独立行走,仅能扶走,不能使用短句表达,不愿意与他人交流,存在言语理解障碍,不能完成简单指令性动作,反应迟钝,可辨别陌生人,但是不能分辨家人角色。
该例患儿入院当天体格检查结果:生命体征平稳,体重为13.0 kg(低于同龄正常儿童2.5个标准差),身高为90.9 cm(低于同龄正常儿童3个标准差),头围为44 cm(低于同龄正常儿童3个标准差),全身无皮疹,浅表淋巴结未触及肿大(图1A)。双眼睑无浮肿,双侧瞳孔等大、等圆,直径约为3 mm,对光反射灵敏;双耳耳廓大,咽部无充血,双侧扁桃体无肿大,双肺呼吸音清,未闻及干湿啰音。上中切牙未见异常(图1B)。心率为116次/min,心律齐,心音有力,各瓣膜听诊区未闻及杂音,腹部平软,肋下未触及肝、脾,肠鸣音正常,肛门外生殖器外观发育正常,尿道口无红肿,脊柱、四肢无畸形,关节活动自如,手指较纤细,无通贯掌,四肢肌力正常,但是肌张力低(图1C)。神经系统查体未见异常。辅助检查结果:白细胞计数为10.6×109/L,中性粒细胞计数为1.33×109/L;乳酸为2.1 mmol/L;血氨为57 μmol/L;游离三碘甲腺原氨酸(free triiodothyronine,FT3),游离甲状腺素(free thyroxine,FT4)与促甲状腺激素(thyroid-stimulating hormone,TSH)甲状腺功能3项检查均正常;肝、肾功能与电解质,以及免疫球蛋白(immunoglobulin,Ig)A、IgG、IgM、补体C3、补体C4和总IgE等免疫功能6项检查均未见异常;CD20+B细胞在淋巴细胞中所占比例为22.10%;17α-羟孕酮为2.226 nmol/L;胰岛素样生长因子为1 117.0 ng/mL,胰岛素样生长因子结合蛋白为32.86 μg/mL;骨碱性磷酸酶为35 μg/L,25-羟维生素D为60.7 nmol/L;单纯疱疹病毒IgM抗体为0.69 AU/mL,风疹病毒IgM抗体为0.28 AU/mL,弓形虫IgM抗体为0.92 AU/mL,巨细胞病毒IgM抗体为0.32 AU/mL;遗传代谢病筛查结果提示瓜氨酸增高;尿液气相色谱-质谱联用仪分析结果提示,尿液中存在大量4-羟基苯乙酸。0~6岁儿童发育检查结果:适应性能力为极重度发育迟缓,大运动能力为中度发育迟缓,精细动作为重度发育迟缓,语言功能为极重度发育迟缓,社交能力为极重度发育迟缓。听性脑干反应(auditory brain-stem responses,ABR)检查结果:左侧听觉传导通路损伤(疑为颅内段损伤),右侧听觉传导通路正常,双侧听觉反应阈值可疑范围。双眼视力、眼底及视觉诱发电位(visual evoked potential,VEP)检查均未见异常。脑电图正常。头颅MRI平扫结果:双侧额部脑外间隙稍增宽。全脊柱正侧位X射线摄片结果:右侧轻度髋发育不良伴右髋关节半脱位可能,腰椎轻度左侧弯(疑为双髋关节不对称所致),其余脊柱未见异常。心电图及心脏、泌尿系统、阴囊彩色多普勒超声检查均未见异常。染色体核型分析:46,XY;遗传性疾病大家系全外显子组检测:携带VPS13B基因2个杂合病理性突变,分别遗传自其母亲和父亲,该基因突变可导致Cohen综合征,多为常染色体隐性遗传。
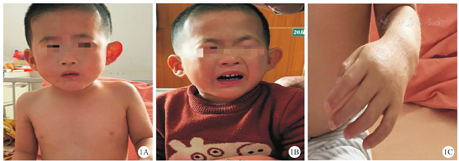
患儿2(先证者),男性,患儿1胞弟,1岁4个月,因"精神运动发育迟缓8+个月",2019年1月1日于中山大学孙逸仙纪念医院儿科门诊就诊。病史采集结果:患儿系G2P2,晚孕期出现胎动减少,足月儿,经阴道自然分娩娩出,出生体重为2 600 g,身长为43.0 cm,出生时无窒息、抢救史,出生时Apgar评分为10分,生后即出现黄疸(血清胆红素值不详),于当地医院接受住院治疗,生后人工喂养。该例患儿分别于8、9、10个月龄时及1岁3个月时,罹患肺炎。精神运动发育迟缓,具体表现为:4+个月龄可抬头,10+个月龄时可独坐,1岁时能爬,于本院就诊时可扶站数分钟,会无意识发出"baba、mama、a、zai"等音,反应迟钝,可辨别陌生人,但是不能分辨家人角色。
该例患儿入院当天体格检查结果:生命体征平稳,体重为9.5 kg(低于同龄正常儿童1.2个标准差),身高为76.0 cm(低于同龄正常儿童1.9个标准差),头围为42 cm(低于同龄正常儿童3个标准差),全身无皮疹,浅表淋巴结未触及肿大。双眼睑无浮肿,双侧瞳孔等大、等圆,直径约为3 mm,对光反射灵敏;双耳耳廓大,咽部无充血,双侧扁桃体无肿大,双肺呼吸音清,未闻及干湿啰音;心率为118次/min,心律齐,心音有力,各瓣膜听诊区未闻及杂音,腹部平软,肋下未触及肝、脾,肠鸣音正常,肛门外生殖器外观发育正常,尿道口无红肿,脊柱及四肢无畸形,关节活动自如,手指较纤细,四肢肌力正常,但是肌张力低。神经系统查体未见异常。辅助检查结果:白细胞计数为7.8×109/L,中性粒细胞计数为1.5×109/L;肝、肾功能,电解质,血氨、血乳酸,FT3、FT4与TSH,骨碱性磷酸酶与25-羟维生素D,胰岛素样生长因子与胰岛素样生长因子结合蛋白,单纯疱疹病毒、风疹病毒、弓形虫、巨细胞病毒IgM抗体,17α-羟孕酮,IgA、IgG、IgM、补体C3、补体C4与总IgE,以及遗传代谢病筛查等,均未见明显异常;CD20+B细胞在淋巴细胞中所占比例为29.43%。0~6岁儿童发育检查结果:适应性能力为极重度发育迟缓;大运动能力为中度发育迟缓;精细动作为重度发育迟缓;语言功能为中度发育迟缓;社交能力为重度发育迟缓。双耳ABR正常;VEP检查:右眼F-VEP P2波潜伏期稍延迟,振幅正常;左眼F-VEP P2波潜伏期未见延迟,振幅正常。脑电图正常;头颅MRI平扫未见异常;全脊柱正侧位X射线摄片未见明显异常;心电图及心脏、泌尿系统、阴囊彩色多普勒超声检查均未见异常;染色体核型分析未见明显异常。遗传性疾病大家系全外显子组检测:携带VPS13B基因2个杂合病理性突变,分别遗传自其父亲和母亲,该基因突变可导致Cohen综合征,多为常染色体隐性遗传。
患儿1、2父亲,44岁。病史采集结果:3岁会说叠词,约3岁6个月能讲短语,可正常交流,语言理解无障碍;运动发育未见明显异常。体格检查结果:步态正常,四肢肌力、肌张力均正常。辅助检查结果:血常规、甲状腺功能3项检查均未见异常。遗传性疾病大家系全外显子组检测:携带VPS13B基因1个杂合病理性突变,患儿1、2的其中1个VPS13B基因病理性突变与此基因突变相同。
患儿1、2母亲,34岁。病史采集结果:语言、运动发育未见明显异常。体格检查结果:步态正常,四肢肌力、肌张力均正常。辅助检查结果:血常规、甲状腺功能3项检查均未见异常。遗传性疾病大家系全外显子组检测:携带VPS13B基因1个杂合病理性突变,患儿1、2的其中1个VPS13B基因病理性突变与此基因突变相同。
患儿1、2表叔,40岁。病史采集结果:3个月龄时可抬头,6个月龄时可独坐,2岁7个月时可独立行走,在此之前不会爬行和扶站;2岁时能叫"爸爸、妈妈",2岁6个月前四肢肌力正常,但是四肢肌张力低;约3岁6个月时能讲短语,可正常交流,言语理解无障碍。体格检查结果:步态正常,四肢肌力、肌张力均正常。辅助检查结果:血常规、甲状腺功能3项检查均未见异常。遗传性疾病大家系全外显子组检测:未检测到与患儿1、2相同的VPS13B基因位点突变。
患儿1、2及其父母与表叔基因检测结果显示,患儿1、2均发生VPS13B[8q22 NM_017890.4 Intron50 c.9259-1G>C][8q22 NM_017890.4 Exon57 c.11104_11105de1]基因突变,见表1;患儿1、2父亲发生VPS13B[8q22 NM_017890.4 Exon57 c.11104_11105de1]基因突变;患儿1、2母亲发生VPS13B[8q22 NM_017890.4 Intron50 c.9259-1G>C]基因突变;患儿1、2表叔进行VPS13B基因c.9259-1G>C位点及c.11104_11105de1位点验证的结果显示,未检测到该突变位点。

患儿1、2遗传性疾病大家系全外显子组检测结果
患儿1、2遗传性疾病大家系全外显子组检测结果
| 基因 | 区带 | 参考序列 | 突变位点 | cDNA水平改变 | 蛋白质水平改变 | 突变状态 | 突变分类 | 患儿父母基因突变检测结果 | |
|---|---|---|---|---|---|---|---|---|---|
| 父亲 | 母亲 | ||||||||
| VPS13B | 8q22 | NM_017890.4 | Intron50 | c.9259-1G>C | p.? | 杂合 | 病理性 | 未检测到 | 杂合携带 |
| VPS13B | 8q22 | NM_017890.4 | Exon57 | c.11104_11105del | p.(Lys3702fs) | 杂合 | 病理性 | 杂合携带 | 未检测到 |
VPS13B 8q22 NM_017890.4 Intron50 c.9259-1G>C p.?突变为剪切突变,该剪切位点突变可导致所编码的蛋白质正常功能丧失,人类基因突变数据库(Human Gene Mutation Database,HGMD)(http://www.hgmd.cf.ac.uk/ac/index.php)目前尚未见该剪切突变报道,但是可见大量其他剪切突变报道;ESP6500siv2_ALL数据库(http://evs.gs.washington.edu/EVS)、千人基因组(1000g2015aug_ALL)(https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes)和dbSNP147数据库(https://www.ncbi.nlm.nih.gov/projects/SNP)等,亦均未见该剪切突变报道。生物信息学软件初步分析结果显示,该剪切突变可明显影响mRNA剪切。因此认为,该突变为病理性突变。
VPS13B 8q22 NM_017890.4 Exon50 c.11104_11105del p.(Lys3702fs)突变为移码突变,可导致所编码的蛋白质自第3 702位氨基酸赖氨酸开始发生移码,并导致蛋白质翻译提前终止,进而导致其所编码的蛋白质发生截短,从而丧失其正常功能。HGMD数据库目前尚未见该移码突变报道,但是可见大量其他移码突变;ESP6500siv2_ALL数据库、千人基因组(1000g2015aug_ALL)和dbSNP147数据库亦均未见该移码突变报道。因此认为,该突变为病理性突变。
根据本研究设定的文献检索策略进行检索的结果显示,共计检索到31篇Cohen综合征相关文献,其中,国内报道为2篇、国外报道为29篇,共计涉及138例Cohen综合征患儿。这138例患儿中,年龄为1~5岁,男性为62例(44.9%),女性为76例(55.1%)。其主要临床表现包括:发育迟缓为136例(98.5%),小头畸形为78例(56.5%),人中短为121例(87.7%),喇叭样鼻子为98例(71.0%),下颌畸形为79例(57.2%),睑裂下斜为20例(14.5%),有过度社交行为者为77例(55.8%),高度近视为62例(44.9%),睫毛浓密为30例(21.7%),躯干性肥胖为84例(60.9%),关节过度伸展为77例(55.8%),四肢纤细为130例(94.2%),视网膜营养不良为54例(39.1%),肌张力低下为121例(87.7%),中性粒细胞减少为130例(94.2%)。发生VPS13B基因病理性突变者为88例(63.8%)。
1973年,Cohen首次报道2例Cohen综合征患儿,系亲姐弟,临床表现为肥胖、肌张力减退、精神缺陷及颅面、眼和肢体异常[1],这2例患儿后来被Carey与Hall[2]证实为罕见染色体遗传性疾病,由位于染色体8q22.2上的空泡蛋白分类13同源物B(vacuolar protein sorting 13 homolog B,VPS13B,又被称为COH1)基因突变的常染色体隐性遗传所致。VPS13B是一种跨膜蛋白,被认为在细胞内囊泡介导的蛋白质运输和分类中发挥作用,并且在眼睛、血液系统和中枢神经系统发育与功能中发挥作用[3,4,5,6]。迄今为止,对于Cohen综合征的诊断标准,尚无统一共识,尤其是对于年龄较小患儿,确诊极为困难。目前,Cohen综合征最常用的诊断标准参考Kolehmainen等[4]提出的标准,即发育迟缓、小头畸形、典型Cohen综合征特殊面容、四肢纤细伴躯干性肥胖、过度社交行为、关节过度伸展、高度近视和(或)视网膜营养不良、中性粒细胞减少等8项临床表现中,若符合6项及以上者,即可诊断为Cohen综合征;若符合5项及以下临床表现者,可诊断为Cohen-like综合征。
Cohen等首次报道的2例Cohen综合征患儿,存在围生期胎动减少[1]。文献报道,多达50% Cohen综合征患儿发生围生期胎动减少[2,3],并且多数患儿为足月儿,但是出生体重和身长为正常新生儿的10%~25%[5]。肌张力减退是Cohen综合征患儿的显著临床表现,可以导致婴儿期出现显著呼吸困难和喂养困难[1,3,6,7]。本研究纳入的2例Cohen综合征患儿在围生期出现明显胎动减少,亦均为足月儿,但是生后未出现呼吸困难和喂养困难,与其他文献报道[1,3,6,7]不一致,这可能与1岁前患儿肌张力减退不明显有关。虽然这2例患儿出生体重和身长均低于同龄正常儿童,但是这并非Cohen综合征的基本临床表现。文献报道,Cohen综合征患儿于青少年时期可能发展为臀部肥胖。然而,Limoge等[10]建议将Cohen综合征患儿"臀部肥胖"改为"异常脂肪分布",因为Cohen综合征患儿的腰围、臀围虽然通常较正常儿童增加,但是其人体质量指数多处于正常范围。该研究结果亦显示,Cohen综合征患儿躯干脂肪积聚增加的原因,是缺乏VPS13B的前脂肪细胞分化为储脂细胞增加,在分化早期,细胞对胰岛素的反应增强,导致特异性脂肪基因加速表达。这提示,Cohen综合征患儿在生长发育中,应做到科学饮食及适量运动,以改善体质,预防肥胖。
Kivitie-Kallio与Norio[3]研究结果显示,多数Cohen综合征患儿2~5岁时仍然不能独立行走。该病患儿1~5岁时会出现语言发育障碍,甚至部分患儿6岁时仍然不能表述完整句子[3]。Cohen综合征患儿均伴有一定程度智力障碍,高达22%患儿伴有严重智力低下[3]。Cohen综合征患儿亦伴有社交障碍,最常见表现为使用非语言交流和言语理解障碍[11]。部分Cohen综合征患儿也可能具有孤独症谱系障碍患者相关表现[12,13]。本研究纳入的2例Cohen综合征患儿均以"精神运动发育迟缓"为主诉就诊。目前,临床对于Cohen综合征患儿的诊治,通常采用物理治疗,通过提高腰部、四肢肌力,促进运动发育,加强言语训练,采取小牛血清去蛋白注射液等营养神经,前列地尔改善循环,促进大脑发育,以改善运动和语言发育迟缓[14]。
Cohen综合征诊断标准之一为典型的面部特征,包括小头畸形、眼裂下斜、头发与眉毛浓密、发际线低、上中切牙突出、上颌骨发育不全、小下颌,张嘴高窄腭弓,耳突出不良,高鼻梁、喇叭样鼻子[3,6]。除了上中切牙突出外,患儿还可能出现牙周早期崩解、广泛的牙槽骨丢失等[6,15]。但是,另有文献报道,Cohen综合征患儿6岁前,临床上很难识别出明显的面部特征[16,17,18]。这与本研究结果相似。本研究纳入的2例患儿,除小头畸形、耳突出不良外,尚未见其他典型面部特征,这可能与该2例患儿年龄尚小有关。
此外,本研究纳入的患儿1行VEP检查未见异常,而患儿2的VEP检查结果示右眼F-VEP P2波潜伏期稍延迟,振幅正常;左眼F-VEP P2波潜伏期未见延迟,振幅正常。Cohen综合征患儿视力呈逐渐恶化趋势,若早期出现进展性高度近视,可于2岁前进行视力矫正;但是于20岁左右,若该病患者视野渐进性收缩,可导致视觉功能恶化[19,20]。该病成年患者多于40岁前出现明显视力损害,但是仅少数患者发生失明[3]。该病患儿近视眼主要为屈光型,由于角膜、睫状体和虹膜发育不全和萎缩,导致虹膜和带状松弛及球形晶体所致[19]。Cohen综合征患儿视网膜营养不良呈渐进性改变,视觉最终可能局限于光感[18]。文献报道,早期矫正视力缺陷,如矫正屈光不正或斜视,对患儿仍具有积极影响[21,22,23]。然而,目前仍然缺乏有效治疗方法阻断色素性视网膜病变的进展。这提示,对于Cohen综合征患儿,应定期进行眼科检查,以评估屈光不正和(或)视网膜营养不良情况。
本研究2例患儿病史采集结果提示,其出生后均多次罹患肺炎,血常规结果提示2例患儿均伴中性粒细胞减少症。由此推测,患儿可能存在先天性中性粒细胞减少症,而这与患儿容易反复感染密切相关,与文献报道一致[3,24]。Cohen综合征患儿多存在先天性中性粒细胞减少症,但是通常属于轻至中度、非周期性和非致命性[3,24]。虽然合并先天性中性粒细胞减少症的Cohen患儿,可能不伴严重细菌感染,但是可能发生反复感染、口腔溃疡、慢性或者复发性牙龈炎[24],使用重组人粒细胞集落刺激因子,可纠正中性粒细胞减少症[12,24]。
多数Cohen综合征患儿四肢纤细。肌张力减退通常在新生儿期即可发现,并且在1岁时症状十分明显,进展到后期可能出现肌肉痉挛[3]。本研究2例患儿全脊柱正侧位X射线摄片均提示右侧轻度髋发育不良并右髋关节半脱位,腰椎轻度左侧弯,这与Jones等[6]文献报道一致。Jones等[6]研究结果显示,Cohen综合征患儿可表现为各种肌肉、骨骼畸形,包括肘外翻、膝外翻、扁平足外翻或内凸、脊柱侧凸、韧带松弛和关节活动过度,多继发于肌张力减退。Cohen综合征患儿还可能伴有单一的腕掌侧横纹,大、小鱼际发育不全,轻度并指畸形,第一脚趾和第二脚趾间隙宽,以及腰椎前凸。
对Cohen综合征患儿神经系统查体,可发现患儿运动不协调或"笨拙"、肌腱反射减弱和肌张力减退[3,6],以及小脑发育不全[24]。文献报道,对Cohen综合征患儿排除其他精神发育迟缓原因后,再行MRI检查结果显示,Cohen综合征患儿的胼胝体可能较正常儿童增大[3,27]。但是,癫痫发作和脑电图异常,并不是Cohen综合征患儿的典型临床表现[1,3]。本研究纳入的2例患儿均未见癫痫发作,头颅MRI和脑电图检查均未见异常,这说明癫痫发作和脑电图异常,并非诊断Cohen综合征的必要条件。本研究中,患儿1生后心脏彩色多普勒超声提示房间隔缺损、动脉导管未闭,而本次入院复查未见心脏缺陷,房间隔缺损和动脉导管未闭已愈合。但是,亦有文献报道,部分Cohen综合征患儿存在心脏缺陷,随着年龄增长,逐渐出现左心室功能下降、瓣膜缺陷(如二尖瓣松弛和二尖瓣关闭不全)、血管缺陷、心脏收缩杂音ST段异常(ST段压低与T波倒置)、原发性高血压和肺动脉高压[3,28,29,30]。因此,临床应注意随访Cohen综合征患儿的心脏彩色多普勒超声及心电图检查。
目前,在Cohen综合征患儿中已发现22种不同VPS13B致病性基因突变,而本研究发现了新的VPS13B致病位点,在HGMD、ESP6500siv2_ALL数据库、千人基因组(1000g2015aug_ALL)和dbSNP147数据库中均尚未见报道,这丰富了HGMD来自中国人群的遗传信息。患儿1、2表叔的病史采集结果显示,其亦存在精神运动发育迟缓,2.5岁前四肢肌张力低,虽然未检测到与患儿1、2相同的VPS13B基因位点突变,但是未能排除存在VPS13B基因其他位点突变可能。El Chehadeh等[31]研究结果显示,所有VPS13B基因突变患儿或伴有脉络膜视网膜营养不良,或伴有中性粒细胞减少症;该研究还评估了Kolehmainen等[4]制定的Cohen综合征诊断标准,认为该诊断标准对于Cohen综合征诊断的敏感度和特异度分别为100%和77%。
综上所述,Cohen综合征属于极为罕见的常染色体隐性遗传性疾病,目前仍然缺乏对该病的充分认识,常误诊为脑性瘫痪。本研究纳入的2例患儿最终依据基因检测结果确诊为Cohen综合征。这提示,对于特殊面容、生长发育落后、高度近视、弱视和色素性视网膜病变伴中性粒细胞减少症的患儿,应考虑Cohen综合征的可能性,并进行基因检测,予以及时确诊。对于Cohen综合征患儿的治疗,应采取积极、规范的康复训练措施,可有效改善其运动发育落后问题,并需长期随访、追踪其并发症发生情况。
所有作者均声明不存在利益冲突

























