
探讨子宫内膜癌(EC)患者预后相关差异基因筛选,并构建其预后预测模型。
在癌症基因组图谱(TCGA)数据库(https://portal.gdc.cancer.gov/)中,以"Uteri" "TCGA-UCEC" "transcriptome profiling" "gene expression quantification and HTSeq-FPKM"为关键词,检索EC患者和正常女性受试者的子宫内膜组织RNA-seq微阵列基因表达数据及其相关临床信息。本研究检索时间设定为TCGA数据库建库至2021年1月15日。选择最终符合本研究纳入标准的542例EC患者与35例正常女性受试者为研究对象。本研究基于TCGA数据库的EC差异基因患者的预后预测模型构建步骤为:①利用R语言微阵列数据的线性模型(LIMMA)包,对TCGA数据库的RNA-seq微阵列基因表达数据进行差异基因分析,筛选影响EC发生、发展的候选差异基因。②利用Kaplan-Meier法、LASSO算法回归、单因素Cox比例风险回归分析法,对EC患者生存相关差异基因进行筛选。采用多因素Cox比例风险回归分析法,确定EC患者预后相关差异基因。③构建EC差异基因的EC患者预后预测模型。④利用survival受试者工作特征(ROC)曲线软件包,检测该预测模型的准确性,并绘制列线图。
①在本组EC患者中,共计发现466个EC差异基因,其中上调基因为179个,下调基因为287个。②在本组EC患者的96个EC生存相关差异基因中,7个为预后相关差异基因,包括孕激素受体(PGR)、sushi重复含蛋白质X连锁(SRPX)、γ-谷氨酰水解酶(GGH)、分泌球蛋白家族2A成员1(SCGB2A1)、胰岛素样生长因子结合蛋白5(IGFBP5)、细胞周期蛋白依赖性激酶抑制剂2A(CDKN2A)、神经调节素U(NMU)基因。对这7个差异基因的单因素Cox比例风险回归分析结果显示,其均为EC患者预后影响因素(P<0.05)。多因素Cox比例风险回归分析结果显示,GGH、IGFBP5、CDKN2A差异基因,均为影响EC患者预后的独立危险因素(P<0.05),若EC患者GGH、IGFBP5、CDKN2A差异基因表达水平越高,则患者预后越差。③建立EC患者总体生存(OS)期预测模型为:ln[h(t,X)/h0(t)]=1.300xGGH+1.200xIGFBP5+1.200xCDKN2A。其中,h(t,X):受试者在t时刻的风险率函数,h0(t):受试者在t时刻的基准风险率函数,即xGGH、xIGFBP5、xCDKN2A均为0时的风险率函数,xGGH、xIGFBP5、xCDKN2A分别表示GGH、IGFBP5、CDKN2A差异基因表达水平。④采用上述预测模型,对研究组患者的生存风险进行评分,并按照其中位风险评分,进一步将其分为高危亚组(n=271,风险评分高于中位评分)与低危亚组(n=271,风险评分低于中位评分),并且低危亚组OS期显著长于高危亚组,差异有统计学意义(χ2=33.000,P<0.001),对该模型预测EC患者OS期的ROC曲线分析结果显示,曲线下面积(AUC)为0.700(95%CI:0.673~0.751,P<0.001),同时构建的Nomogram列线图,可定量预测EC患者1、3、5年OS率。
构建的GGH、IGFBP5和CDKN2A差异基因的EC患者预后预测模型,可为临床预测EC患者预后及寻找相应靶向治疗药物提供数据支持。


版权所有,未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别申明,本刊刊出的所有文章不代表中华医学会和本刊编辑委员会的观点。
本刊为电子杂志,以光盘形式出版。本册应读者需求按需印刷,随光盘免费赠阅。光盘如有质量问题,请向编辑部调换。
子宫内膜癌(endometrial cancer,EC)是女性生殖道3大恶性肿瘤之一,发病率及其导致的患者病死率呈逐年上升趋势[1]。EC是美国女性第4大常见癌症,其新发病例占女性癌症患者的7%,90%该病患者的组织病理类型为内膜样腺癌,其余为黏液性、浆液性腺癌及透明细胞癌等[2]。对于复发或晚期EC患者,以及具有临床侵袭性的EC患者的临床预后常不佳[3]。因此,寻找EC预后相关分子标志物,尽早对高危EC患者进行靶向治疗,是延长EC患者总体生存(overall survival,OS)期的重要策略[4]。
生物信息学分析在癌症研究中已被广泛应用,克服了由于不同平台和小样本数据导致的分析结果不一致或分析受限难题,收集大量数据,为寻找肿瘤患者预后相关分子标志物,以及构建肿瘤相关预测模型提供重要研究手段[5]。美国癌症基因组图谱(The Cancer Genome Atlas, TCGA)计划由美国国家癌症研究所(National Cancer Institute,NCI)和国家人类基因组研究所(National Human Genome Research Institute,NHGRI)发起,目前TCGA数据库中收录了很全面的癌症基因组数据,包括突变、拷贝数变异、mRNA表达、miRNA表达、甲基化数据等[6],并且完成33种不同癌症类型的数据,总数据量达到2.5 PB[7]。笔者拟对来自TCGA数据库的542例EC患者与35例正常女性受试者的子宫内膜组织基因表达数据,利用R语言(R 4.0.2)的微阵列数据的线性模型(linear models for microarray data,LIMMA)包,筛选EC差异基因[8],并结合其生存资料,筛选预后相关基因,构建其OS期预测模型,为高危EC患者靶向治疗提供参考。现将研究结果报道如下。
本研究检索关键词为"Uteri" "TCGA-UCEC" "transcriptome profiling" "gene expression quantification and HTSeq-FPKM" ;检索时间设定为TCGA数据库建库至2021年1月15日。从TCGA数据库(https://portal.gdc.cancer.gov/)中,下载548例EC患者与35例正常女性受试者的子宫内膜组织RNA-seq微阵列基因表达数据及其相关临床信息。选择符合本研究纳入标准的542例EC患者与35例正常女性受试者为研究对象。将其分别纳入研究组与对照组。其中,研究组患者年龄为(63.9±11.2)岁;国际妇产科联盟(International Federation of Gynecology and Obstetrics,FIGO)临床分期为Ⅰ~Ⅳ期者分别为338例(62.4%)、51例(9.4%)、123例(22.7%)与30例(5.5%);组织病理分级为G1~G4者分别为97例(17.9%)、121例(22.3%)、313例(57.8%)与11例(2.0%)。
本研究受试者纳入标准[9]:①分泌期中期的子宫内膜组织mRNA转录组数据存储于TCGA数据库的EC患者和正常女性受试者;②EC患者和正常女性受试者临床资料完整者。
对TCGA数据库研究组与对照组的转录组数据进行log2标准化后,采用R语言(R 4.0.2)的LIMMA包,对TCGA数据库研究组与对照组的转录组数据进行贝叶斯检验,筛选EC差异基因,并使用pheatmap包绘制研究组患者的差异基因热图[10]。本研究EC差异基因筛选标准:对研究组与对照组受试者倍性变化(fold change,FC)取对数,若|log2FC|≥1,错误发现率(false discovery rate,FDR)<0.05的基因,则筛选为EC差异基因。其中,FC表示研究组患者子宫内膜差异基因表达量与对照组正常女性受试者子宫内膜差异基因表达量的比值。FDR表示所有发现的差异基因中,实际并非差异基因所占比例。
采用Kaplan-Meier法,分析EC患者的差异基因对其OS期的影响,按照研究组患者的差异基因的中位表达水平,将其进一步分为高风险亚组(差异基因表达水平高于研究组的中位表达水平者)和低风险亚组(差异基因表达水平低于研究组的中位表达水平者),对2个亚组患者的OS期进行比较,将差异有统计学意义(P<0.05)的差异基因,作为EC患者生存相关基因。对研究组患者筛选的EC生存相关基因,使用R语言的survival包进行LASSO回归后[11],进一步进行单因素Cox比例风险回归分析。再对单因素Cox比例风险回归分析结果中差异有统计学意义(P<0.05)的差异基因,进一步进行双向逐步回归法多因素Cox比例风险回归分析,筛选预测EC患者预后相关基因,并构建EC患者OS期预测模型。最后使用受试者工作特征(receiver operating characteristic,ROC)曲线软件包,检测该预测模型对EC患者预后预测准确性[12],并绘制列线图,以便临床更便捷、客观预测患者预后情况[13]。本研究所有统计学检验采用双侧检验,以P<0.05表示差异有统计学意义。
对EC差异基因的筛选结果显示,自研究组患者中,共计发现466个EC差异基因,其中上调基因为179个,下调基因为287个。研究组患者EC差异基因分层聚类热图,见图1。
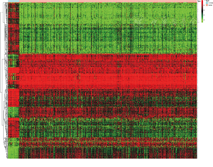
注:红色表示EC差异基因高表达(上调),绿色表示EC差异基因低表达(下调)。EC为子宫内膜癌
对研究组患者生存相关差异基因筛选结果显示,自466个EC差异基因中,筛选出96个EC生存相关差异基因,包括7个预后相关差异基因:孕激素受体(progesterone receptor, PGR),sushi重复含蛋白质X连锁(sushi repeat containing protein X-linked, SRPX),γ-谷氨酰水解酶(gamma-glutamyl hydrolase,GGH),分泌球蛋白家族2A成员1(secretoglobin family 2A member 1, SCGB2A1),胰岛素样生长因子结合蛋白5(insulin like growth factor binding protein 5, IGFBP5),细胞周期蛋白依赖性激酶抑制剂2A(cyclin dependent kinase inhibitor 2A, CDKN2A),神经调节素U(neuromedin U, NMU)差异基因(图2A、图2B)。对这7个差异基因的单因素Cox比例风险回归分析结果显示,其均为EC患者预后影响因素(P<0.05)。进一步对PGR、SRPX、GGH、SCGB2A1、IGFBP5、CDKN2A、NMU差异基因进行双向逐步回归法多因素Cox比例风险回归分析结果显示,GGH、IGFBP5、CDKN2A差异基因高表达,均为影响EC患者预后的独立危险因素(P<0.05),而PGR、SCGB2A1、SRPX与NMU差异基因高表达,均非EC患者独立影响因素(P>0.05);建立的EC患者OS期预测模型为ln[h(t,X)/h0(t)]=1.300xGGH+1.200xIGFBP5+1.200xCDKN2A。其中,h(t,X):受试者在t时刻的风险率函数,h0(t):受试者在t时刻的基准风险率函数,即xGGH、xIGFBP5、xCDKN2A均为0时的风险率函数,xGGH、xIGFBP5、xCDKN2A分别表示GGH、IGFBP5、CDKN2A差异基因表达水平,见图2C。EC患者预后相关差异基因的单因素与多因素Cox比例风险回归分析结果,见表1。
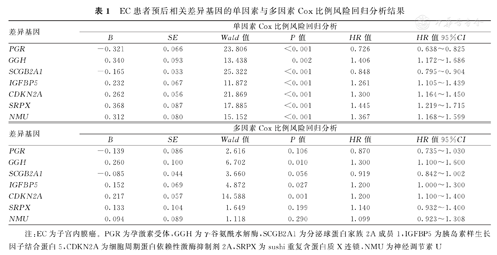
EC患者预后相关差异基因的单因素与多因素Cox比例风险回归分析结果
EC患者预后相关差异基因的单因素与多因素Cox比例风险回归分析结果
| 差异基因 | 单因素Cox比例风险回归分析 | |||||
|---|---|---|---|---|---|---|
| B | SE | Wald值 | P值 | HR值 | HR值95%CI | |
| PGR | -0.321 | 0.066 | 23.806 | <0.001 | 0.726 | 0.638~0.825 |
| GGH | 0.340 | 0.093 | 13.438 | 0.002 | 1.406 | 1.172~1.686 |
| SCGB2A1 | -0.165 | 0.033 | 25.322 | <0.001 | 0.848 | 0.795~0.904 |
| IGFBP5 | 0.232 | 0.067 | 11.872 | <0.001 | 1.261 | 1.105~1.439 |
| CDKN2A | 0.262 | 0.056 | 21.869 | <0.001 | 1.300 | 1.164~1.450 |
| SRPX | 0.368 | 0.087 | 17.885 | <0.001 | 1.445 | 1.219~1.715 |
| NMU | 0.312 | 0.080 | 15.152 | <0.001 | 1.367 | 1.168~1.599 |
| 差异基因 | 多因素Cox比例风险回归分析 | |||||
|---|---|---|---|---|---|---|
| B | SE | Wald值 | P值 | HR值 | HR值95%CI | |
| PGR | -0.139 | 0.086 | 2.616 | 0.106 | 0.870 | 0.735~1.030 |
| GGH | 0.260 | 0.100 | 6.702 | 0.010 | 1.300 | 1.100~1.600 |
| SCGB2A1 | -0.085 | 0.044 | 3.660 | 0.056 | 0.919 | 0.842~1.002 |
| IGFBP5 | 0.152 | 0.069 | 4.872 | 0.027 | 1.200 | 1.000~1.300 |
| CDKN2A | 0.217 | 0.057 | 14.588 | 0.001 | 1.200 | 1.100~1.400 |
| SRPX | 0.133 | 0.104 | 1.649 | 0.199 | 1.140 | 0.932~1.400 |
| NMU | 0.094 | 0.089 | 1.118 | 0.290 | 1.099 | 0.923~1.308 |
注:EC为子宫内膜癌。PGR为孕激素受体,GGH为γ-谷氨酰水解酶,SCGB2A1为分泌球蛋白家族2A成员1,IGFBP5为胰岛素样生长因子结合蛋白5,CDKN2A为细胞周期蛋白依赖性激酶抑制剂2A,SRPX为sushi重复含蛋白质X连锁,NMU为神经调节素U
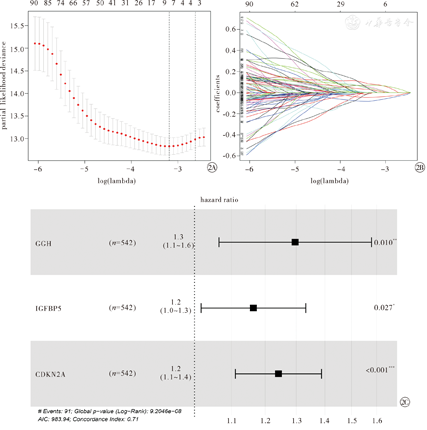
注:EC为子宫内膜癌。GGH为γ-谷氨酰水解酶,IGFBP5为胰岛素样生长因子结合蛋白5,CDKN2A为细胞周期蛋白依赖性激酶抑制剂2A。log(lambda)为惩罚系数,partial likelihood deviance为部分似然偏差,coefficients为系数,hazard ratio为风险比
采用建立的EC患者OS期预测模型,对研究组患者生存风险进行评分,并根据其中位评分,将其进一步分为高危亚组(n=271,评分高于中位评分)与低危亚组(n=271,评分低于中位评分)。使用R语言的survival ROC软件包检测该模型对研究患者预后预测准确性的分析结果显示,低危亚组患者的OS期显著长于高危亚组,并且差异有统计学意义(χ2=33.000,P<0.001)(图3A);对该模型预测EC患者OS期的ROC曲线的曲线下面积(area under curve, AUC)为0.700(95%CI: 0.673~0.751,P<0.001)(图3B);该模型可对高、低危EC患者进行区分(图3C,图3D,图3E),并可通过Nomogram列线图的差异基因表达评分定量预测EC患者1、3、5年OS率(图4)。
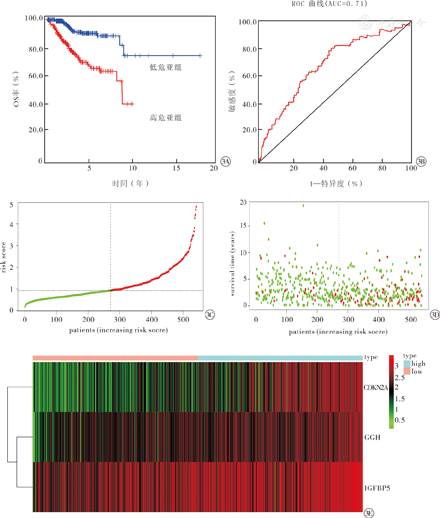
注:EC为子宫内膜癌。OS为总体生存,ROC曲线为受试者工作特征曲线,AUC为曲线下面积。GGH为γ-谷氨酰水解酶,IGFBP5为胰岛素样生长因子结合蛋白5,CDKN2A为细胞周期蛋白依赖性激酶抑制剂2A。patients (increasing risk score)为患者(风险评分增加),risk score为风险评分,survival time (years)为生存时间(年)
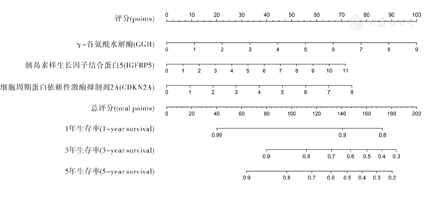
注:EC为子宫内膜癌。OS为总体生存
EC是常见妇科恶性肿瘤,近年该病发病率呈增高趋势[14]。EC具有分子异质性,不同患者预后不同,目前认为Ⅰ型和Ⅱ型非雌激素依赖型EC可能与PTEN、P16、P53等基因突变相关[15]。但是,迄今为止,临床对EC患者的诊断和预后预测,仍然主要根据患者肿瘤形态学特征,如组织病理学类型、肿瘤分级和肿瘤细胞侵袭性等[16]。目前,FIGO临床分期和组织病理学类型,是临床对EC患者治疗方案选择和预后评估最常用依据[17,18]。FIGO临床分期虽然对晚期EC患者预后判断较准确,但是对于早期EC患者预后判断效果则一般,这表明FIGO临床分期对于准确预测EC患者预后具有局限性[19]。基于TCGA数据库的挖掘和分析,鉴别肿瘤相关生物标志物,建立预后预测模型,已被证明是一种有效预测肿瘤患者预后的方法[20]。
本研究中,通过对TCGA数据库RNA-seq微阵列基因表达数据的分析,鉴定了EC相关差异基因。再采用Kaplan-Meier法,筛选EC生存相关差异基因,并对其进行LASSO回归、单因素和多因素Cox比例风险回归分析,筛选出3个EC预后相关差异基因GGH、IGFBP5和CDKN2A。最后,本研究构建由这3个差异基因组成的EC患者OS期预测模型,用于预测EC患者预后,这为进一步探索EC发病机制提供研究方向,对建立新的EC患者预后风险分类和寻找相应靶向治疗药物奠定基础。本研究建立的EC患者OS期预测模型中,GGH、IGFBP5和CDKN2A差异基因是EC患者预后不良影响因素,EC患者GGH、IGFBP5、CDKN2A差异基因表达水平越高,其预后越差。既往研究证明,GGH是参与叶酸代谢的酶,与邻近癌旁组织相比,GGH在乳腺癌和ERG阴性前列腺癌组织中高表达,并且此类患者预后较差[21,22]。GGH亦为口腔鳞状细胞癌患者预后不良影响因素[23]。IGFBP5已被证明在子宫内膜、卵巢、前列腺等组织中表达[24]。Naciff等[25]研究表明,IGFBP5与EC细胞Ishikawa(ISK)雌激素信号通路表达相关;Miyake等[26]证明,IGFBP5在体外可促进前列腺癌细胞生长,并可加速前列腺癌动物模型肿瘤进展。研究表明,卵巢癌患者血清IGFBP5水平在复发时显著高于初始诊断,这提示IGFBP5参与卵巢癌进展[27]。CDKN2A在各种肿瘤中经常发生突变或缺失,并通过CDK4和p53对细胞周期进行调节。大量研究证明,CDKN2A高表达与EC患者预后不良相关[28,29,30]。Su等[31]临床荟萃分析结果显示,CDKN2A高甲基化可能是早期EC癌变的标志,其高表达往往提示EC患者预后较差。
2013年,TCGA将EC分为极点超突变、微卫星不稳定性高突变、拷贝数低和拷贝数高4种类型[32]。2015年,Stelloo等[33]依据EC患者临床病理因素,将其分为DNA聚合酶ε(DNA polymerase epsilon,POLE)突变型,微卫星不稳定型,无特异性分子变异型与TP53突变型,前2类EC患者预后良好,而后2类EC患者预后较差。Zhang等[34]研究发现,CCNB2、CDC20、BUB1B、UBE2C、AURKB、FOXM1、NCAPG、RRM2、TPX2、DLGAP5、CDCA8、CDC45、MKI67、BUB1、KIF2C基因表达水平与EC患者预后相关。He等[35]发现,L1CAM、MUC15、CDON、IGSF9B、PCDH9、PCDHB1基因表达水平,可以对EC患者OS期和无进展生存期进行良好预测。本研究根据TCGA数据库,对542例EC患者转录组数据进行分析,构建GGH、IGFBP5和CDKN2A差异基因对EC患者OS期预测模型,采用该模型对542例EC患者的OS曲线分析结果显示,高危患者OS期显著短于低危患者,并且差异有统计学意义(P<0.05),AUC为0.700(95%CI:0.673~0.751,P<0.001),该模型可以定量预测EC患者1、3、5年OS率,将其与患者病理类型结合,可及时识别高危患者,对EC患者OS期具有良好预测能力。
本研究通过对TCGA数据库中EC患者RNA-seq微阵列基因表达数据进行分析,筛选出与EC发生、发展相关的差异基因,并建立GGH、IGFBP5、CDKN2A差异基因对EC患者OS期预测模型,可为临床预测EC患者预后及寻找相应靶向治疗药物提供数据支持。由于本研究是基于TCGA数据库中数据集构建的预测模型,尚需进一步收集多中心、大样本临床数据验证该模型的有效性和可靠性。
所有作者均声明不存在利益冲突

























