
探讨二酰甘油O-酰基转移酶(DGAT)1基因突变所致先天性腹泻与肠病(CODEs)患儿的临床及基因突变特点。
选择2020年12月,于华中科技大学同济医学院附属武汉儿童医院诊治的1例DGAT1基因突变所致CODEs患儿(以下以示区别称其为本例患儿)为研究对象。采用回顾性分析方法,分析本例患儿的临床病例资料,检索国内外数据库中关于DGAT1基因突变CODEs患儿相关研究的中、英文文献,总结该病患儿的临床及基因突变特点。本研究通过本院伦理委员会审查(审批文号:2021R045-E01)。监护人对本例患儿的诊治知情同意。
①本例患儿为女性,生后50 d时,因呕吐、腹泻就诊,入院查体发现其伴营养不良,实验室检查主要为低蛋白血症及高甘油三酯血症,基因检测结果为DGAT1基因exon 1:c.133delG(p.Asp45Thrfs*22)移码突变,为纯合突变,其父母均为DGAT1基因该位点突变携带者。本例患儿经低脂奶粉喂养及对症支持等治疗26 d后,临床治愈出院。其出院诊断为,CODEs(DGAT1基因纯合突变)。②文献检索获得关于DGAT1基因突变CODEs患儿相关研究文献为10篇,纳入CODEs患儿为29例,加上本例患儿共计纳入30例的分析结果如下。这30例患儿均于婴儿期起病(19例在新生儿期起病),首发主要表现为腹泻(26/30,86.7%),呕吐(19/30,63.3%)及营养不良(20/30,66.7%);同时合并低蛋白血症(15/30,50.0%)和高甘油三酯血症(9/30,30.0%);DGAT1基因突变位点主要包括g.13827T>C、c.629_631delCCT、c.314C>T及c.1202G>A等,主要突变类型为剪接突变、移码突变、错义突变等。对其治疗均以对症支持治疗为主,同时辅以低脂或无脂饮食喂养,大部分患儿临床症状改善。
本组CODEs患儿均由于DGAT1基因突变所致,多于新生儿期起病,主要临床表现为持续、严重、慢性腹泻与呕吐及营养不良,辅以低脂或无脂饮食可改善其临床症状。


未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计,除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。本刊为电子期刊,以网刊形式出版。
先天性腹泻与肠病(congenital diarrhea and enteropathies,CODEs)是一类罕见病,由于肠道上皮功能障碍等所致,以持续、严重、慢性腹泻为主要表现的先天性疾病,主要为单基因突变所致[1]。这类疾病多在新生儿期起病,除腹泻外,部分患儿还可存在不同程度肠外表现,如低蛋白血症、水肿、生长发育迟缓及电解质紊乱等;由于该病起病早、病情重,治疗困难,导致患儿死亡率较高[1,2]。
二酰甘油O-酰基转移酶(diacylglycerol O-acyltransferase,DGAT)1基因突变所致CODEs,又被称为腹泻7型(MIM:615863),以新生儿期起病的持续呕吐、腹泻、营养不良、喂养困难及生长发育迟缓为主要特征,目前国内文献对其报道不多。本研究对1例DGAT1基因突变所致CODEs患儿的临床表现及基因突变特点进行总结,并结合相关文献进行文献复习,旨在为临床医师诊治该病提供临床经验。现将研究结果报道如下。
选择2020年12月,于华中科技大学同济医学院附属武汉儿童医院住院治疗的1例CODEs患儿为研究对象(以下以示区别称其为本例患儿)。本研究通过本院伦理委员会审查(审批文号:2021R045-E01)。监护人对本例患儿的诊治知情同意。
目前对CODEs患儿的诊断,尚无相关指南或金标准。本研究根据相关文献及临床经验,对该病患儿的诊断标准如下[1,2]。①婴儿早期出现腹泻,多于新生儿期起病;②腹泻持续时间>2周;③主要表现为严重腹泻(血便、水样便及脂肪泻等)及呕吐等消化道表现,部分患儿合并多种肠外表现(高脂血症、营养不良等);④大部分患儿基因检测结果提示IL10RA、CYBB、FOXP3、DGAT1等基因突变,且突变为致病性;⑤排除变态反应性疾病、新生儿坏死性小肠结肠炎及短肠综合征等获得性疾病所致腹泻。
以"DGAT1基因" "先天性腹泻病" "腹泻7型" "DGAT1 mutation" "congenital diarrhea" "congenital diarrhea type 7"等为关键词,对中国知网数据库、万方数据知识服务平台及PubMed文献数据库中,关于DGAT1基因突变所致CODEs的相关文献进行检索。本次文献检索的检索年限设定为上述数据库建库至2020年12月。
本例患儿为女性,生后50 d时,因"呕吐1个月余,加重伴喂养困难7 d",于2020年12月就诊于华中科技大学同济医学院附属武汉儿童医院。其系G1P1,39+5周胎龄时经阴道分娩,出生体重为3 250 g,羊水量少,伴羊水粪染(程度不详)。其生后因考虑"出生窒息",于当地医院新生儿科住院7 d(诊治情况不详),出院后给予混合喂养,每次奶量约为60 mL,3~4 h喂食一次。于当地医院出院当日即出现呕吐,呕吐物为奶汁,1~2次/d,每次量不等;大便呈黄色稀糊状,6~7次/d,不伴腹胀、便血及抽搐等,未予特殊治疗,生后30 d时体重为3 000 g。本次就诊的7 d前,呕吐症状加重,3~5次/d,摄入奶量减少,约为30 mL/次,6~8次/d,精神欠佳、小便量减少;同时伴大便次数增加,呈黄色蛋花样,量较多,无黏液脓血等,约10次/d;近10 d体重下降400 g;无发热、抽搐、意识障碍及咳嗽等。患儿父母均体健,非近亲婚配,未提供家族遗传代谢性疾病史。入院拟诊为:呕吐原因待查、重度营养不良。
体重为2 500 g,身高为51.6 cm,头围为34 cm,发育、营养不良;神志清楚,消瘦貌,肤色苍白,皮肤弹性差,皮下脂肪菲薄,无皮疹、水肿,浅表淋巴结无肿大;头颅形态正常,前囟未闭约为2 cm×2 cm,眼睑正常,巩膜无黄染,瞳孔等大、等圆,直径为2.5 mm,对光反射正常;唇干燥,咽无充血;心、肺、腹及四肢检查正常;原始反射可正常引出。
本例患儿入院后实验室检查结果如下。①以下项目基本正常:血液分析、肾功能、血糖、心肌酶、甲状腺功能、促肾上腺皮质激素水平正常,血清抗核抗体为阴性。②肝功能结果:球蛋白、白蛋白及前白蛋白,均低于正常值,分别为15.4 g/L、26.2 g/L、114.2 g/L;其余项目基本正常。③血电解质结果:血清K+为2.86 mmol/L、Mg2+为0.53 mmol/L、Na+为134.9 mmol/L、P为0.55 mmol/L、Ca2+为1.8 mmol/L,均低于正常值。④大便检查:大便常规正常,隐血弱阳性,大便轮状病毒及腺病毒抗原学检测均为阴性。⑤血氨浓度正常,血乳酸为3.79 mmol/L高于正常值。⑥血气分析:除了脉搏血氧饱和度为91%(正常)外,血pH值为7.31、动脉血氧分压为66 mmHg(1 mmHg=0.133 kPa)、动脉血二氧化碳分压为28 mmHg、HCO3-为14 mmol/L及碱剩余为-12,均低于正常值。⑦免疫全套:免疫球蛋白(immunoglobulin, Ig)M为0.25 g/L(正常),IgA为0.71 g/L高于正常值,补体3<0.18 g/L、补体4<0.07 g/L及IgG为2.85 g/L,则均低于正常值。⑧血脂全套:总胆固醇为1.83 mmol/L与脂蛋白a为2.6 mmol/L,均正常;高密度脂蛋白胆固醇为0.57 mmol/L与低密度脂蛋白胆固醇为0.89 mmol/L,均低于正常值;甘油三酯为1.64 mmol/L高于正常值。⑨血清胰岛素水平为1.45 μIU/mL与血清25羟基-维生素D为6.14 ng/mL,均低于正常值。⑩血氨基酸酰基肉碱串联质谱及尿有机酸气相色谱检测,均未见明显异常。
本例患儿入院时影像学检查结果如下。①全消化道X射线造影检查未见明显器质性病变。②肝、胆、脾超声检查结果示肝、脾未见明显异常,空腹胆囊约为3.1 cm×0.5 cm(长径×短径),壁薄、光滑,囊内可见1个强回声光团,直径约为0.7 cm,考虑胆囊结石。③颅脑MRI检查未见明显异常,DWI未见弥散受限表现。
征得患儿监护人同意后,对患儿及其父母进行全外显子基因检测及拷贝数变异(copy number variants,CNV)检测结果显示,患儿DGAT1基因exon 1:c.133delG(p.Asp45Thrfs*22)移码突变(图1),为纯合突变,其父母均为该位点突变基因携带者。根据笔者文献检索认为,此突变尚未见文献报道。该基因突变编码蛋白预测为有害,根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)的《ACMG遗传变异分类标准与指南(2015年)》[3]确认,该变异被判定为致病性变异;患儿CNV检测结果为阴性。
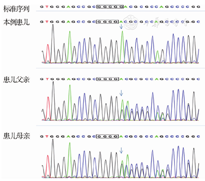
注:CODEs为先天性腹泻与肠病,DGAT1为二酰甘油O-酰基转移酶1
根据本研究设定的检索策略,符合条件的中文文献为1篇[4],英文文献为9篇[5,6,7,8,9,10,11,12,13],研究共计纳入29例DGAT1基因突变所致CODEs患儿。对本例患儿及上述29例CODEs患儿的临床病例资料进行分析如下。
入院后,对本例患儿进行深度水解奶喂养,同时补液、静脉营养、静脉输注白蛋白及丙种球蛋白等对症支持治疗后,患儿呕吐逐渐好转,但是腹泻好转不明显。根据其临床表现,结合全外显子基因检测结果,被诊断为CODEs(DGAT1基因纯合突变)。确诊该病后,调整患儿为低脂奶粉喂养,其大便次数逐渐减少,呈稀糊状,2~3次/d。患儿住院26 d后好转出院。4个月龄后逐渐添加米粉、粥类等辅食,避免进食脂类含量较高食物,患儿耐受可。出院后持续门诊随诊中,2~3个月/次。目前患儿1岁7个月龄,无呕吐、腹泻,身高为79.0 cm、体重为9.0 kg,分别位于同性别、年龄健康儿童的3%~10%、3%;患儿扶物可站立,Gesell发育量表评分为76分,能发"爸爸""妈妈"等叠音字。
这30例患儿均根据临床表现与基因检测结果被诊断CODEs。①一般临床资料:男性患儿为17例、女性13例;除2例未描述出生胎龄外,22例为足月儿,6例为早产儿;除3例未提供发病年龄外,其余患儿发病年龄为生后至3个月龄,其中19例患儿在新生儿期起病。②主要症状:首发表现主要为腹泻(26/30,86.7%),呕吐(19/30,63.3%)及营养不良(20/30,66.7%)。③实验室检查:主要表现为低蛋白血症(15/30,50.0%)和高甘油三酯血症(9/30,30.0%)。④治疗及结局:由于多数患儿存在低蛋白血症,因此多采用静脉注射白蛋白和(或)丙种球蛋白支持治疗,并调整为低脂或无脂饮食喂养;大部分患儿临床症状改善。③基因检测:突变位点主要包括g.13827T>C、c.629_631delCCT、c.314C>T及c.1202G>A等,主要突变类型包括剪接突变、移码突变、错义突变等。24例患儿为单一位点突变,6例患儿存在2处位点复合突变。该病存在较明显的家族遗传倾向,16例患儿的致病基因来源于父亲和(或)母亲,其中包括基因突变位点及类型完全一致的2对双胎患儿(病例13与14、23与24)。本例患儿DGAT1基因突变位点为exon 1:c.133delG(p.Asp45Thrfs*22),纯合突变,移码突变,该突变位点既往参考文献尚未见报道,来源于其父亲和母亲,其父、母均为正常表型。对30例CODEs患儿DGAT1基因突变分析结果,见表1。
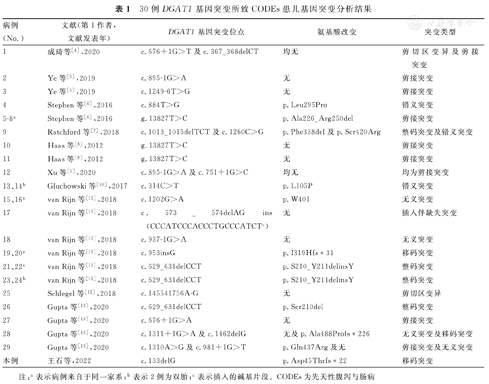
30例DGAT1基因突变所致CODEs患儿基因突变分析结果
30例DGAT1基因突变所致CODEs患儿基因突变分析结果
| 病例(No.) | 文献(第1作者,文献发表年) | DGAT1基因突变位点 | 氨基酸改变 | 突变类型 |
|---|---|---|---|---|
| 1 | 成琦等[4],2020 | c.676+1G>T及c.367_368delCT | 均无 | 剪切区变异及剪接突变 |
| 2 | Ye等[5],2019 | c.895-1G>A | 无 | 剪接突变 |
| 3 | Ye等[5],2019 | c.1249-6T>G | 无 | 剪接突变 |
| 4 | Stephen等[6],2016 | c.884T>G | p.Leu295Pro | 错义突变 |
| 5-8a | Stephen等[6],2016 | g.13827T>C | p.Ala226_Arg250del | 剪接突变 |
| 9 | Ratchford等[7],2018 | c.1013_1015delTCT及c.1260C>G | p.Phe338del及p.Ser420Arg | 整码突变及错义突变 |
| 10 | Haas等[8],2012 | g.13827T>C | 无 | 剪接突变 |
| 11 | Haas等[8],2012 | g.13827T>C | 无 | 剪接突变 |
| 12 | Xu等[9],2020 | c.895-1G>A及c.751+1G>C | 均无 | 均为剪接突变 |
| 13、14b | Gluchowski等[10],2017 | c.314C>T | p.L105P | 错义突变 |
| 15、16a | van Rijn等[11],2018 | c.1202G>A | p.W401 | 无义突变 |
| 17 | van Rijn等[11],2018 | c.573_574delAG ins(CCCATCCCACCCTGCCCATCTc) | 无 | 插入伴缺失突变 |
| 18 | van Rijn等[11],2018 | c.937-1G>A | 无 | 无义突变 |
| 19、20a | van Rijn等[11],2018 | c.953insG | p.I319Hfs*31 | 移码突变 |
| 21、22a | van Rijn等[11],2018 | c.629_631delCCT | p.S210_Y211delinsY | 整码突变 |
| 23、24b | van Rijn等[11],2018 | c.629_631delCCT | p.S210_Y211delinsY | 整码突变 |
| 25 | Schlegel等[12],2018 | c.145541756A-G | 无 | 剪切区变异 |
| 26 | Gupta等[13],2020 | c.629_631delCCT | p.Ser210del | 整码突变 |
| 27 | Gupta等[13],2020 | c.676+1G>A | 无 | 剪接突变 |
| 28 | Gupta等[13],2020 | c.1311+1G>A及c.1462delG | 无及p.Ala488Profs*226 | 无义突变及移码突变 |
| 29 | Gupta等[13],2020 | c.1310A>G及c.981+1G>T | p.Gln437Arg及无 | 剪接突变及无义突变 |
| 本例 | 王石等,2022 | c.133delG | p.Asp45Thrfs*22 | 移码突变 |
注:a表示病例来自于同一家系;b表示2例为双胎;c表示插入的碱基片段。CODEs为先天性腹泻与肠病
根据发病机制不同,可将CODEs分为肠道上皮转运障碍、肠道上皮酶及代谢异常、肠道上皮极性及其细胞信号转导异常、肠道内分泌细胞异常、免疫功能异常相关肠病5种类型,其中以免疫功能异常相关肠病最常见[1,14]。目前,对大部分CODEs已明确致病基因,而且种类较多,包括IL-10、IL-10RA、CYBB、NCF1、FOXP3、XIAP、DGAT1、PCSK1等,其中IL-10、CYBB等致病基因主要引起患儿免疫功能异常相关肠病,DGAT1基因突变可导致肠道上皮代谢障碍,如腹泻、生长发育迟缓等[1,2,14,15]。
DGAT1基因位于8号常染色体,转录和翻译后形成DGAT1蛋白,属于膜表达酰基转移酶家族成员之一,在甘油三酯合成过程中发挥重要作用[16]。当DGAT1基因缺陷导致DGAT1蛋白生成减少后,机体自身甘油三酯合成减少;同时,DGAT1蛋白参与脂质代谢、信号转导及小肠脂肪吸收等多种代谢途径,在脂类物质的形成、吸收和转运中发挥重要作用[16,17,18,19]。2012年,Haas等[8]首先报道DGAT1基因突变所致CODEs家系,揭示该基因与CODEs的关系,但是对CODEs的具体发病机制尚不明确。该病发病机制可能为二酰基甘油或脂肪酸在机体过量堆积所致的毒性作用,也可能为肠上皮细胞内Na+/葡萄糖协同转运蛋白1(Na+-dependent glucose transporter 1,SGLT1),二肽酶IV(dipeptidyl peptidase-IV,DPPIV)和Na+/H+交换体3(Na+/H+ exchanger 3,NHE3)等各种酶减少所致[14,15]。Takemoto等[17]使用DGAT1抑制剂处理小鼠后,小鼠出现水样腹泻,由于肠道上皮细胞内脂类物质代谢障碍,过量堆积的脂肪酸的细胞毒性作用,导致其肠道上皮功能紊乱,继而腹泻及体重增加受限。
DGAT1基因突变所致CODEs,又被称为腹泻7型,多致患儿生后2周内起病,主要症状包括腹泻、呕吐、营养不良等,均存在不同程度低蛋白血症,部分患儿合并高甘油三酯血症及低蛋白血症水肿,治疗过程中需要及时补充白蛋白[4,5,6,7,8,9,10,11,12,13]。本例患儿主要表现为呕吐、腹泻及营养不良,辅助检查结果提示低蛋白血症、高甘油三酯血症,与上述检索获取文献报道一致。同时,本例患儿血清25羟基-维生素D水平明显降低,提示患儿维生素D缺乏,这与van Rijn等[11]报道一致,但是导致维生素D缺乏的具体机制尚迄今不明确。对CODEs患儿进行消化内镜检查,肉眼未见明显特异表现,小肠标本病理检查结果可见微绒毛局部空泡化或萎缩等[2,4,5]。本例患儿因监护人拒绝,而未行消化内镜检查,患儿的确诊依据为临床表现及基因检测结果,出院诊断考虑DGAT1基因突变所致CODEs。DGAT1基因突变位点主要包括g.13827T>C、c.629_631delCCT、c.314C>T及c.1202G>A等,主要突变类型包括剪接突变、移码突变、错义突变等,本例患儿该基因突变位点为exon 1:c.133delG(p.Asp45Thrfs*22),为纯合突变、移码突变,既往尚未见文献报道该突变类型,来源于其父亲和母亲,但是父母均为正常表型,符合常染色体隐性遗传特点。
由于对DGAT1基因突变所致CODEs尚无有效、特异性治疗措施,目前治疗方案主要为对症支持治疗,包括肠外营养支持、保持机体内环境稳定、低脂饮食喂养等[1,2,18,19]。多数患儿在确诊前,由于不能排除变态反应性疾病所致腹泻等原因,而采用深度水解配方奶、氨基酸配方奶喂养,并联合肠外营养加强支持治疗,但是对腹泻、呕吐及营养不良的改善效果欠佳,故在确诊该病后,选择低脂配方奶或无脂配方奶喂养,患儿耐受度较好,上述症状逐渐减轻,但是考虑到低脂配方奶或无脂配方奶不能补充足量脂类物质,部分患儿逐渐调整为中链脂肪酸配方奶,或低脂辅食喂养,患儿耐受情况尚可[4,5,6,7,8,9,10,11,12,13]。在明确诊断CODEs并进行针对性饮食调整后,多数患儿生长发育逐渐好转,但是较同龄健康儿仍表现为营养不良。本研究文献复习涉及的29例患儿中,5例死亡,其余24例正持续随访中。
随着分子生物学进步及新一代测序技术不断革新,包括DGAT1基因在内的多种基因异常所致的CODEs致病机制得以阐明。由于该病起病早、进展快,治疗难度大,因此早期识别、早期干预至关重要。对于呕吐、腹泻症状严重及持续时间长,同时存在营养不良等的患儿,建议尽早进行基因检测,明确病因后采取针对性治疗措施改善患儿预后。
所有作者均声明不存在利益冲突

























