
探讨非典型良性家族性新生儿癫痫(BFNE)患儿的临床特征、家系调查及基因突变分析,并总结我国KCNQ基因突变所致癫痫患儿的遗传学特点。
选择2021年2月,于四川大学华西第二医院确诊的1例延迟在3个月龄癫痫发病的BFNE患儿(先证者)为研究对象。回顾性分析其临床表现、家系调查及基因检测结果等。以"KCNQ基因""癫痫"为中文关键词,以"KCNQ gene""seizures""epilepsy""convulsion"为英文关键词,对中国知网、万方数据知识服务平台(年限设定为上述数据库建库至今),以及Google Scholar、PubMed数据库(年限设定为2000年1月至2020年12月)进行检索,总结我国KCNQ基因突变癫痫患儿的遗传学特点。本研究遵循的程序符合2013年新修订的《世界医学协会赫尔辛基宣言》要求。与本例患儿监护人签署临床研究知情同意书。
①本例患儿为3个月龄女婴,因"反复抽搐1 d"入院。其头颅MRI未见异常;脑电图结果提示多灶性癫痫波发放。调查本例患儿家系3代8人发现,先证者(本例患儿)及其父亲、祖父均发生婴儿期惊厥,非新生儿期发病,其父亲、祖父的BFNE均于1岁左右消失。对本例患儿经左乙拉西坦抗癫痫治疗至9个月龄后抽搐发作停止,随访至1岁1个月亦未再发。②本例患儿及其父亲、祖父基因检测结果显示,均存在KCNQ 2基因2号外显子存在c.373dupG(p.A125fs)杂合突变。与BFNE相关。③文献复习结果:根据本研究设定的检索策略,共检索到129篇国内关于KCNQ基因突变致癫痫发作的相关研究文献,纳入研究的中国患儿为129例,加上本例患儿共计130例。这130例患儿中,72例的起病年龄为0~28 d,56例为29 d至2岁,2例>2岁;KCNQ 2、3基因突变各为119例与11例;随访105例,51例患儿合并智力/发育障碍,其中KCNQ 2、3基因突变各为50例与1例,5例患者死亡,均为KCNQ 2基因突变,其余患儿智力/发育正常。本组130例KCNQ基因突变致癫痫患儿中,108例接受药物治疗患儿的治疗有效率为69.4%(75/108),105例接受随访患儿的死亡率为4.8%(5/105)。
KCNQ 2基因c.373dupG(p.A125fs)突变,可能为本例患儿家系BFNE发病的分子遗传学致病机制。国内KCNQ基因突变致癫痫患儿以KCNQ 2基因突变为主,患儿多于2岁内起病,并且KCNQ 2基因突变癫痫患儿相较于KCNQ 3基因突变患儿预后更差。


未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计,除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。本刊为电子期刊,以网刊形式出版。
良性家族性新生儿癫痫(benign familial neonatal epilepsy,BFNE)是婴儿期早发性癫痫中最具遗传异质性的癫痫综合征,呈常染色体显性遗传,外显率约为85%[1]。BFNE的特征是在生后第2~3天出现短暂局灶性癫痫发作或全面性强直性发作,伴呼吸暂停或阵挛性运动,每天数次至数十次不等;该病为自限性病程,可在几周或几个月内自然缓解,BFNE患儿通常预后良好,但是仍有约15% BFNE患儿出现反复惊厥发作,甚至进展为发育性癫痫性脑病[1,2,3]。目前认为BFNE是由编码电压门控K+通道亚基的KCNQ 2、3基因突变所致,这些基因是构成神经细胞M型K+通道的分子基础,基因缺陷时M电流减少是导致新生儿BFNE发病的主要机制[3]。本研究对1例生后缺乏典型BFNE临床表型,而延迟在3个月龄发病的BFNE患儿进行回顾性分析,并对近年国内外该病的相关文献进行复习,旨在探讨我国KCNQ基因突变致癫痫患儿的临床及遗传学特征。现将研究结果报道如下。
选择2021年2月,于四川大学华西第二医院确诊的1例BFNE患儿为研究对象,确诊时其月龄为3个月。本研究遵循的程序符合2013年新修订的《世界医学协会赫尔辛基宣言》要求。患儿监护人均签署临床研究知情同意书。
本研究根据国际抗癫痫联盟(International League Against Epilepsy,ILAE)分类和术语委员会(https://www.epilepsydiagnosis.org)制定的BFNE诊断标准,其特征包括生后第1~28天癫痫发作,12个月内发作自行缓解,发作间期神经学检查结果正常,头颅MRI结果无异常,KCNQ基因检测提示致病性基因突变,则可确诊为BFNE [2]。
抽取患儿及其父母静脉外周血3 mL,并置于含乙二胺四乙酸(ethylene diamine tetraacetic acid)抗凝管中保存待测,对患儿及其父母、祖父母、外祖父母的基因全外显子检测及患儿全基因组拷贝数变异(copy number variation, CNV)分析。采用序列捕获技术将全基因组所有外显子区域DNA捕获,并富集后进行高通量测序基因分析(武汉康圣达医学检验所),目标区域覆盖度为99.8%,目标区域平均深度为114.64×,目标区域平均深度>20×为99.1%。测序数据与1000 Genomes Project、ESP6500、gnomAD等数据库提供的人类基因组hg19参考序列进行比对,突变位置根据相应的转录本进行注释,预测突变的软件包括Scale-Invariant Feature Transform(SIFT)、Polyphen-2等生物信息分析软件,突变致病性判定查阅人类基因突变数据库(Human Gene Mutation Database,HGMD)(http://www.hgmd.org), ClinVar等数据库及参考文献,基因与疾病的关系,通过在线人类孟德尔遗传(Online Mendelian Inheritance in Man, OMIM)数据库(https://omim.org/)注释判断。根据2015年美国医学遗传与基因组会(American College of Medical Genetics and Genomics,ACMG)的遗传突变分类标准与指南,将突变分级为致病、可能致病、临床意义未明、疑似良性和良性。
以"KCNQ基因" "癫痫"为中文关键词,以"KCNQ gene""seizures""epilepsy""convulsion"为英文关键词,在中国知网、万方数据知识服务平台(检索年限设定为上述数据库建库至今),以及Google Scholar、PubMed数据库中(检索年限设定为2000年1月至2020年12月),检索关于KCNQ基因突变所致我国癫痫发作患儿的相关研究文献。
本例患儿为女性,就诊时月龄为3个月,2021年2月因"反复抽搐1 d"于四川大学华西第二医院住院治疗。患儿系G1P1,足月经剖宫产术娩出,出生后无窒息史,出生体重为3.3 kg,生长发育同正常同龄儿。入院前抽搐表现为意识丧失,呈强直阵挛性发作,1 d内反复发作10次,每次发作持续2~3 min,发作后精神差,进食减少,不伴发热、呕吐、腹泻等;父母非近亲结婚。入院体格检查:生命体征平稳,神清,前囟未闭、平软(1.5 cm×1.5 cm),颈阻呈阴性,竖头欠稳,四肢肌张力正常,病理性反射不能引出。常规头颅MRI检查结果未见异常;脑电图结果提示睡眠期多灶性棘波、尖波、棘慢波、尖慢波发放;脊髓穿刺脑脊液检查正常;血尿串联质谱及气相色谱检测呈阴性。入院后予以口服左乙拉西坦抗癫痫治疗,初始剂量为10 mg/(kg·d)×2次/d,每周加量10 mg/kg,加量调整直至获得最佳疗效和耐受性,最大剂量≤60 mg/(kg·d)。经治疗后患儿癫痫发作停止,好转出院。出院后持续左乙拉西坦治疗至9个月龄,对其随访至1岁1个月,癫痫未再发作,生长发育可。
对本例患儿家系的3代8人进行调查发现,其中受累者为3人,分别为先证者(Ⅲ2)、先证者父亲(Ⅱ3)及先证者祖父(Ⅰ2),其癫痫临床症状基本一致,表现为于婴儿期起病,1岁左右惊厥消失;其余调查对象(5人)无相关临床表现,智力及生长发育评估均正常。本例患儿(先证者)BFNE发病家系图,见图1。
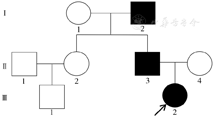
注:BFNE为良性家族性新生儿癫痫。Ⅰ、Ⅱ、Ⅲ分别指家系第1~3代(祖、父、孙3代)。□与○为男、女性未患病者,■与●为男、女性患病者, 为先证者
为先证者
对本例患儿家系3例BFNE患者的基因全外显组进行测序结果发现,本例患儿KCNQ 2基因2号外显子存在c.373dupG(p.A125fs)杂合突变,该突变的染色体位置为chr20:62078113(基因组版本:hg19),与BFNE相关;患儿父亲及祖父KCNQ 2基因2号外显子存在c.373dupG(p.A125fs)杂合突变;其余被调查对象未检测到KCNQ 2基因2号外显子存在c.373dupG(p.A125fs)杂合突变,为野生型(图2)。本例患儿CNV未见明显异常。
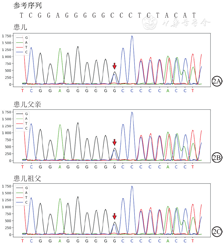
注:BFNE为良性家族性新生儿癫痫
c.373dupG(p.A125fs)突变位于KCNQ 2基因,cDNA序列第373位碱基(G)重复,导致编码区第125位丙氨酸发生移码突变,KCNQ蛋白构象变化,可能影响该蛋白质正常生物学功能。c.373dupG(p.A125fs)突变在HGMD专业版数据库中无收录,Clinvar数据库中无收录。KCNQ基因该位点突变的人群频率无收录,在gnomAD数据库的东亚人群中亦无收录。参考2015年ACMG基因突变解读指南,KCNQ基因该位点突变ACMG证据:可能致病(PVS、PM2)。
出院后,对本例患儿随访至1岁1个月,抽搐未再发作,复查脑电图结果提示正常,智力及体格检查均正常。本例患儿非新生儿期发病,但是结合临床表型及家系遗传基因特点分析,确诊为BFNE。
根据本研究设定的文献检索策略,检索到KCNQ基因突变所致癫痫发作患儿研究的中文文献为21篇[4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24],英文文献为8篇[25,26,27,28,29,30,31,32],纳入129例KCNQ基因突变所致癫痫患儿,均为中国国籍,加上本例患儿,共计130例。根据KCNQ基因突变所致癫痫特点,对这130例患儿性别、发病年龄、诊断、发作类型、药物治疗及疗效、遗传特点等进行总结。
130例患儿的临床诊断中,早发性癫痫性脑病(early-onset epilepsy encephalopathy,EOEE)为41例,BFNE为23例,良性家族性婴儿癫痫(benign familial infantile epilepsy,BFIE)为19例,大田原综合征(Ohtahara syndrome,OS)为13例,婴儿痉挛症(infantile spasm,IS)为9例,电压依赖性K+通道2(potassium voltage-gated channel,subfamily Q,member 2,KCNQ 2)脑病为8例,癫痫性脑病(epileptic encephalopathy,EE)为6例,婴儿癫痫伴游走性局灶性发作(epilepsy of infancy with migrating focal seizures,EMIFS)为3例,良性新生儿癫痫(benign neonatal epilepsy,BNE)为3例,不能分类者为2例,其他诊断如额叶癫痫,全面性癫痫伴热性惊厥附加症(generalized epilepsy with febrile seizures plus,GEFS+),Lennox-gastaut综合征(Lennox-Gastaut syndrome,LGS)各为1例。
①性别:男性患儿为74例、女性为56例,男、女性别比为1.3∶1。②起病年龄:72例(55.4%)为0~28 d,56例(43.1%)为29 d至2岁,2例(1.5%)为>2岁;起病中位年龄3 d(1~730 d)。③发作类型:全面性癫痫发作为52例,部分性发作为47例,不能分类的发作为15例,未描述发作类型为16例。④脑电图特点:多灶放电为31例,暴发抑制为30例,高度失律为12例,正常为11例,局部发电为4例,交替图形为1例,未获得脑电图结果41例。⑤头颅MRI:正常为60例,异常为13例,早期表现为基底节区/脑白质异常,后期表现为脑萎缩及胼胝体改变,未描述头颅MRI检查结果为57例。⑥家族史:有明确家族史者为45例,无家族史者为68例,未描述家族史为17例。
采用单药治疗为60例,包括单用奥卡西平、托吡酯、丙戊酸钠、左乙拉西坦、苯巴比妥等;采用联合治疗方案为50例,包括托吡酯+氨己烯酸、奥卡西平+苯巴比妥、左乙拉西坦+苯巴比妥、托吡酯+苯巴比妥、丙戊酸钠+托吡酯等;未用药治疗为2例;未描述用药方案为18例。治疗有效为75例,无效为33例,未描述治疗疗效为22例。接受随访患儿为105例,其中,智力/发育正常为49例,异常为51例,死亡为5例。
鉴定出的致病性KCNQ基因突变中以KCNQ 2基因最常见(91.5%,119/130),KCNQ 3基因少见(8.5%,11/130),暂未见其他KCNQ基因致癫痫文献报道。5例死亡患儿均源于KCNQ 2基因突变;51例患儿合并智力/发育障碍,其中KCNQ 2、3基因突变分别为50、1例;预后不良分别占KCNQ 2及KCNQ 3基因突变患儿总数的46.2%(55/119)及9.1%(1/11)。
癫痫发作是婴幼儿期患儿神经内科就诊的常见急症,是指由脑部神经元的过度放电引起的一种急性、反复发作、阵发性的大脑功能紊乱,表现为意识、运动、自主神经和精神障碍。长时间或顽固性癫痫发作,对患儿认知及脑功能可造成不利影响,特别是发育期儿童大脑易受癫痫潜在影响,导致患儿发育阻滞或倒退,最终导致其发生发育性癫痫性脑病;除去颅脑外伤、感染、窒息等继发癫痫病因外,部分癫痫属于遗传因素所致[33]。鉴于K+电流的不同亚细胞定位、生物物理特性、调节作用和药理学特性等,电压门控K+通道在调节脑部神经元固有电位及其对不同输入突触响应方面起着关键作用[33]。文献报道,某些K+通道基因突变是导致癫痫发病的首要原因[34]。
电压门控K+通道家族是离子通道家族中最大且最多样化的家族,包含约80个基因,其中KCNQ (Kv7)基因亚家族成员,是电压门控K+通道亚基中最重要的成员之一,Kv7由相同或类似亚基组成四聚体,共编码6个跨膜结构域、单孔环及K+通道a亚基;而编码产物KCNQ蛋白,则广泛存在于心脏、大脑、平滑肌、骨骼肌及感觉器官等组织中,在细胞质、细胞内的细胞器与细胞外隔间离子稳态中起着重要作用。KCNQ (Kv7)亚家族由5个成员组成,分别是KCNQ1~5 (Kv7.1~ 5),每个成员都有不同的表达模式和功能角色。Kv7.1亚基主要在心肌细胞中表达,辅助KCNE1亚基共同构成延迟整流K+电流对心室的再极化,是导致长QT综合征(long QT syndrome,LQTS)的离子通道之一;Kv7.4亚基在耳蜗内感觉外毛细胞中高表达,调节这些细胞的内在兴奋性,在听力中发挥重要作用;Kv7.2、3、5通道亚基主要存在于中枢神经元,包括海马的齿状回和CA1~3区域、新皮质及丘脑的网状核,能异质化形成M电流(M current,IM),IM抑制可使神经元去极化,增加神经元兴奋性,而IM激活,可使细胞膜超极化,从而抵消兴奋性刺激,降低神经元兴奋性[34,35],目前,KCNQ 2、KCNQ 3和KCNQ 5基因突变的致病性,已在部分癫痫综合征中被证实[36]。
本研究文献复习结果显示,130例我国KCNQ基因突变所致癫痫患儿中,主要为KCNQ 2基因突变(119例),而KCNQ 3基因突变少见(11例),未见KCNQ 5基因突变;KCNQ基因突变导致癫痫发病儿童中,<2岁占总人数98.5%(128/130);46.2%(55/119)KCNQ 2基因突变患儿可能经历反复癫痫发作,表现为不同类型的癫痫综合征,遗留严重的神经和智力损伤,部分患儿甚至因惊厥死亡,预后相较于KCNQ 3基因突变患儿的预后更差。二者虽同为Kv7.2/Kv7.3通道改变,但是同一基因的突变体可能导致患儿发生不同癫痫综合征及预后,推断其原因可能与良性及非良性癫痫共享致病基因相关;欧洲人群KCNQ基因突变患儿的癫痫发作类型,绝大多数表现为癫痫部分性发作[30],而文献结果显示中国人群KCNQ基因突变患儿癫痫全面性发作与部分性发作并无显著差别(52∶47)。
虽然BFIE和BFNE均具有婴儿早期出现特发性癫痫发作、无癫痫综合征、精神运动发育正常、发作间期脑电图正常、1岁以内发病但数周或数个月后自行缓解、常染色体显性遗传等特点[37]。但是,由于KCNQ基因突变位点不同,BFIE并不是BFNE的等位基因形式[38]。本例患儿及其家系发病成员惊厥发作均发生在生后数月,非新生儿期发病,不具备典型BFNE的临床表现,似乎不能由BFNE解释,但既往BFNE研究文献已有相关特点报道[39,40]。本例患儿家系全外显子基因检测结果显示,该家族在20号染色体KCNQ 2基因存在c.373dupG突变,导致编码区第125位丙氨酸发生移码突变(p.A125fs),可能导致基因功能丧失,是导致患儿发生BFNE的机制,结合本例患儿临床表型及家系遗传基因特点分析,最终仍确诊为BFNE。这种非典型BFNE与BFIE发作形式一致,表明二者之间可能存在某种重叠或遗传联系,但其具体作用机制尚需进一步研究以证实。
KCNQ 2基因c.373dupG(p.A125fs)突变,可能为本例患儿及其家系成员发生BFNE的遗传学病因,这是首次在中国人群中发现的位点突变,丰富了KCNQ基因突变谱。部分KCNQ 2基因突变BFNE患儿临床表型,可与BFIE患儿表型一致。目前,国内KCNQ基因突变癫痫患儿以KCNQ 2基因突变为主,主要起病于2岁以内,预后与KCNQ基因突变类型及癫痫类型密切相关。
所有作者均声明不存在利益冲突

























