
探讨编码纤维蛋白原(Fg)γ多肽链基因-FGG突变,所致新生儿先天性纤维蛋白原缺乏症(CA)的临床及遗传学特点。
选择2021年5月,青岛大学附属医院新生儿科诊治的1例CA新生儿为研究对象,回顾性分析其临床病例资料。采用单基因病高通量测序技术,检测本例患儿外周血FGG基因,患儿父母进行特定基因位点的Sanger法测序,以明确患儿基因突变来源。对中国知网数据库、万方数据知识服务平台及PubMed数据库中,关于新生儿期起病的FGG基因突变所致CA病例进行检索,总结CA新生儿的临床及遗传学特点。本研究遵循的程序符合2013年修订的《世界医学协会赫尔辛基宣言》要求。监护人对患儿的诊治知情同意,并签署临床研究知情同意书。
①临床资料:本例患儿系女性,生后11.7 h时,因"发现面部淤斑4 h"入院,无发热,不伴其他部位出血等。除面部淤斑外,体格检查无异常。入院时凝血常规检查结果异常,主要为抗凝血酶Ⅲ与血浆Fg含量均较正常值低,分别为31.0%与0.18 g/L;凝血酶原时间与凝血酶时间均较正常值延长,分别为22.3 s与24.0 s。同时,患儿母亲于分娩前发现血浆Fg减少(为0.52 g/L),不伴出血症状。②本例患儿及其母亲外周血检出FGG(4q28|NM_000509.4)基因exon 8:c.1073C>A p.(Ser358Tyr)错义突变、杂合突变,患儿该突变来自其母亲。该突变位点在人类基因变异数据库(HGMD)等基因库中目前未见收录,亦未见文献报道,生物信息学软件预测其有致病可能。③经静脉输注冷沉淀治疗后,患儿症状好转出院,出院诊断为新生儿CA,低出生体重儿。对本例患儿随访至5个月龄,无出血表现。④文献检索结果:仅发现3例新生儿期起病的FGG基因突变所致CA患儿。其中,患儿1、2(患儿1缺乏详细描述,患儿2为女性,诊断时为10 d龄)为FGG基因缺失/移码突变(均为纯合子)所致CA,FGG基因突变分别为g.194delA、c.1096delC,临床症状为脐带断端出血和(或)关节腔积血。患儿3为男性,来自一个CA家系(母亲FGG基因突变纯合子,为先证者),患儿3基因检测为FGG基因c.1073C>G错义突变(杂合子),并且仅表现为轻度出血。
新生儿期起病的CA患儿目前被报道较少,临床对该病认识尚不足。通过Fg相关基因检测对该病进行早期诊断,可预防危及其生命的成年期创伤、术后严重出血等。


未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计,除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。本刊为电子期刊,以网刊形式出版。
先天性纤维蛋白原缺乏症(congenital afibrinogenemia,CA)是常染色体隐性遗传的出血性疾病,其特征为先天性血浆纤维蛋白原(fibrinogen,Fg)缺乏,导致患者不同程度出血[1]。该病的普通人群发病率为1%~2%[2],占罕见出血障碍(先天性凝血因子缺乏)的8%[3]。对CA患者目前尚无有效治疗方法,主要以补充Fg等替代治疗为主[4]。对编码Fg 3种多肽链的基因FGA、FGB、FGG进行检测,可以确诊该病,并可以增强对临床表型的预测[5]。上述3种基因的纯合或复合杂合无效突变,易引起患儿严重出血,而杂合、错义突变患儿出血相关临床表现常不典型[6]。因此,这部分临床表现不典型CA患儿容易被漏诊,以至于在成年期,其易因创伤、手术或妊娠发生严重出血而危及生命。新生儿期精准的分子诊断,不仅可以明确CA病因,还可为家庭提供适当遗传咨询,避免其成年期发生不可控的严重出血。本研究拟对1例CA新生儿的临床病例资料进行分析,旨在加深儿科医师对该病FGG基因突变类型和相关表型的认识,并为临床诊断该病提供相关的遗传缺陷新信息。现将研究结果报道如下。
选择2021年5月,于青岛大学附属医院新生儿科住院治疗的1例CA患儿为研究对象。本研究遵循的程序符合2013年修订的《世界医学协会赫尔辛基宣言》要求。监护人对患儿的诊治知情同意,并签署临床研究知情同意书。
CA患者主要临床表现为不同程度出血。若患儿血浆Fg<0.1 g/L,则可被诊断为先天性无纤维蛋白原血症;若血浆Fg为0.1~1.5 g/L,则被诊断为先天性低纤维蛋白原血症。对患者进行Fg相关基因检测,可明确病因[7]。
抽取患儿及其父母外周静脉血3 mL,采用乙二胺四乙酸抗凝,外送至上海金域检验公司进行基因检测:提取DNA对患儿进行单基因病全外显子及其旁侧内含子高通量测序;采用Sanger法测序,对患儿及其父母进行特定基因位点的基因突变检测,以明确患儿基因突变来源。
以"先天性纤维蛋白原缺乏症" "先天性纤维蛋白原减少症" "先天性无纤维蛋白原血症" "先天性低纤维蛋白原血症" "FGG" "新生儿" "congenital afibrinogenemia" "congenital hypofibrinogenemia"为关键词,对中国知网数据库、万方数据知识服务平台及PubMed文献数据库中,关于新生儿期起病CA患儿的FGG基因突变的相关研究文献进行检索。本次文献检索的检索年限设定为上述数据库建库至2021年10月。
本例患儿为女性,生后11.7 h时,因"发现面部淤斑4 h",于2021年5月7日于青岛大学附属医院新生儿科住院治疗。患儿入院前4 h,家属发现患儿面部淤斑,躯干、四肢等其他部位无出血点及淤斑。患儿无抽搐、激惹,无发热、咳嗽、血尿、便血等其他症状。患儿系G1P1,足月剖宫产术娩出,出生体重为2 420 g,生后1、5、10 min Apgar评分均为10分。其母孕期合并妊娠期糖尿病,通过饮食控制,孕期血糖控制良好。孕母分娩前凝血功能检查显示结果异常,血浆Fg含量为0.52 g/L(正常参考值为1.50~3.50 g/L),而被诊断为低纤维蛋白原血症,静脉输注冷沉淀800 U及Fg 2 g治疗后,复查血浆Fg含量为1.36 g/L,剖宫产术中失血量约为300 mL。患儿入院诊断为:出血原因待查;低出生体重儿。
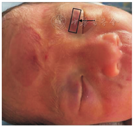
患儿一般情况可。面部散在淤斑,最大为15 mm×5 mm,眼周为著(图1)。躯干、四肢等其他部位皮肤无出血点、淤斑及黄染。患儿心、肺、腹、四肢及神经系统查体无异常。
①患儿血、尿、大便常规,血气分析、生化全套检查项目基本正常,常规血培养结果呈阴性,颅脑超声未见异常。②患儿(11.7 h龄)入院时、静脉输注冷沉淀治疗(3 d龄)时,以及8 d龄时,分别进行凝血常规检查示(以下正常参考值均为健康足月新生儿参考值):抗凝血酶Ⅲ分别为31.0%、54.0%、45.0%(正常参考值为51.0%~75.0%);凝血酶原时间分别为22.3、12.0、12.6 s(正常参考值为13.0~20.0 s);部分凝血活酶时间分别为61.3、61.3、58.2 s(正常参考值为45.0~65.0 s);血浆Fg含量分别为0.18、0.70、0.47 g/L(正常参考值为1.17~2.25 g/L);凝血酶时间分别为24.0、22.0、23.2 s(正常参考值为10.0~16.0 s);D-二聚体分别为400、400、330 ng/mL(正常参考值为0~500 ng/mL)。
患儿及其母亲外周血检出FGG(4q28|NM_000509.4)基因exon 8:c.1073C>A p.(Ser358Tyr)错义突变、杂合突变(图2),其编码蛋白质的第358位氨基酸由丝氨酸(Ser)改变为酪氨酸(Tyr),患儿父亲FGG基因检测结果正常。患儿及其母亲FGG基因该位点突变,在HGMD数据库(http://www.hgmd.cf.ac.uk/)、ESP6500siv2_ ALL(http://evs.gs.washington.edu/EVS/),千人基因组(1000g2015aug_ALL)(https://www.internationalgenome.org/)和dbSNP147数据库(https://www.ncbi.nlm.nih.gov/snp/)均未见收录;在NCBI数据库(https://www.ncbi.nlm.nih.gov/geo/)中进行查询,无该位点突变的数据,并且排除基因多态性可能。由此考虑患儿FGG基因exon 8:c.1073C>A为新的错义突变。对该突变进行生物信息学分析结果显示:蛋白同源性分析显示该蛋白序列在多种脊椎动物中均有较高保守性;Mutation-Assessor软件的突变得分为3.68分,预测该突变具有很高致病性;Polyphen-2_HDIV软件的蛋白序列变异得分为1分,预测该蛋白序列改变可能具有致病性。
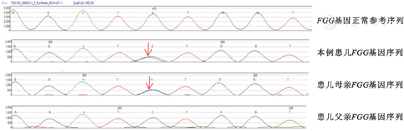
注:CA为先天性纤维蛋白原缺乏症
患儿入院1 d后,对其采取静脉输注冷沉淀25 mL/次×1次/d×3 d,同时给予维生素K1、酚磺乙胺(商品名:止血敏)止血等措施治疗,患儿面部淤斑减轻,全身无新发出血点。患儿住院7 d后,好转出院,出院诊断为:新生儿CA,低出生体重儿。对患儿定期门诊随访至生后5个月龄时,生长发育可,无出血表现,未再进行静脉输注冷沉淀及Fg治疗。
根据本研究设定的检索策略,检索到符合条件的英文文献为3篇[8,9,10],中文文献无。涉及新生儿期起病的FGG基因突变所致CA患儿共计3例。其中,Neerman-Arbez等[8]报道1例CA患儿(患儿1,缺乏性别及发病年龄等信息描述),因基因检测发现FGG基因g.194delA缺失突变(纯合子),而被诊断为CA患儿(父母为近亲结婚),但文献缺少对其他临床资料描述。2020年,Tavasoli等[9]报道的14例CA患者中,仅1例10 d龄新生儿(患儿2,女性),因脐带断端出血和关节腔积血(未详细描述临床表现,故具体积血关节腔不详),基因检测发现FGG基因c.1096delC移码突变(纯合子),而被诊断为CA。Casini等[10]报道在一个CA家系中的1例新生儿(患儿3,男性),基因检测发现FGG基因c.1073C>G(p.Ser358Cys)错义突变,而被诊断为CA患儿。该家系先证者为患儿3的母亲,既有血栓表型,又有严重出血表型,临床表现为内脏血栓形成、鼻出血及月经过多,基因检测结果为FGG基因c.1073C>G(p.Ser358Cys)纯合子。但是,该先证者FGG基因发生相同突变的子代(患儿3,杂合子)的临床出血症状较轻(文献缺少对患儿3其他临床表现详细描述)。
先天性纤维蛋白原疾病,是由Fg相关基因突变所致血浆Fg含量或功能异常,Fg含量异常(Ⅰ型,即CA)包括无纤维蛋白原血症、低纤维蛋白原血症,Fg功能异常(Ⅱ型)包括纤维蛋白原异常血症、低异常纤维蛋白原血症。其中,CA为具有活性的血浆Fg含量减少,为0~1.5 g/L[5]。Fg又被称为凝血因子Ⅰ,是分泌型糖蛋白复合物,分子质量约为340 kDa,在肝脏产生。Fg参与血凝块形成的最后阶段,在机体凝血过程中发挥至关重要作用,Fg在凝血酶激活后,Fg转变为纤维蛋白,纤维蛋白单体形成并自发聚集,随后联合血小板形成稳定血凝块,而达到止血目的[11]。正常成年人血浆Fg含量为1.5~3.5 g/L,Fg在血浆中的半衰期约为4 d[12]。Fg是由Aα、Bβ和γ 3种多肽链组成的二聚体,3种多肽链分别由FGA、FGB、FGG 3种基因编码,3种基因共同位于染色体4q28-4q31约50 kb区域内,其中FGG基因由8.5 kb的10个外显子组成。
1935年,CA被首次被报道,直到1999年,研究人员才首次发现导致该病的FGG基因缺陷[13]。近年来,随着基因检测技术迅速发展,高通量测序技术广泛应用于临床,越来越多Fg相关基因突变类型被发现,而这些突变分散于Fg相关的3种基因,但很少有突变热点。本研究纳入研究患儿的FGG基因c.1073C>A突变,系应用单基因疾病高通量测序技术发现,尚未被HGMD、dbSNP147等数据库收录,这个新的杂合子错义突变,通过患儿临床症状结合生物信息软件推测,考虑为导致该例患儿发生CA的致病原因。
FGG基因杂合子错义突变,可导致CA患儿血浆Fg含量降低,而导致临床出血表现,其可能的机制是这些错义突变多位于γ链高度保守的C-末端球状结构域中的缘故[14]。对Fg功能研究表明,这些位置FGG基因错义突变,可破坏Fg分子结构稳定性,而致Fg六聚体的组装或分泌受损[15]。对仓鼠卵巢细胞的体外实验也证实,γY278H细胞系的Fg水平低,是由于γY278H细胞的Fg虽然可以从肝细胞正常分泌,但随后在血液循环中,却可被纤溶酶加速降解[16]。由此可见,FGG基因的结构稳定,对于维持Fg功能至关重要。
CA患者通常症状较轻,中等水平血浆Fg含量,常足以防止患者自发性出血[1]。但是,严重低Fg (血浆Fg含量<0.5 g/L)或创伤后CA患者,则容易发生大出血[17]。该病的早期诊断困难,患儿常在凝血试验筛查中被发现,其中凝血酶时间指标异常对诊断CA最敏感,但病因诊断仍有赖于Fg相关基因检测。该病基因型与临床表型相关性差,临床表现异质性明显,若结合特异性凝血试验(Fg抗原功能测定),如血栓弹性图或凝血酶生成试验,可更好预测临床表型[18]。本例患儿因家属拒绝而未能完成上述Fg功能测定试验。Sumitha等[19]对27例印度裔纤维蛋白原疾病患者的研究结果显示,无Fg血症患者在生命早期(≤6.5岁)可被诊断,而低Fg血症患者的诊断年龄平均为21岁。因此,对于漏诊的症状较轻低Fg血症患儿,常在成年期因创伤或手术时才被诊断,表现为创伤后重要脏器,包括颅内出血、脾出血、手术出血不止等,罹患该病孕妇可有自发性流产、胎盘早剥等严重产科并发症。血浆Fg含量不足,会导致凝血酶活性增加,从而增加血栓形成风险[20]。
对CA患者早期诊断困难,故为其后期治疗造成困扰,甚至导致终身残疾。文献报道1例父母为近亲结婚婴儿,生后1个月内发生持续长时间脐带断端出血,因为未引起重视,至4个月龄时,家属发现患儿无视物追踪,遂进行眼科及相关实验室检查,发现患儿系血浆Fg水平低导致的双侧玻璃体腔出血而最终引起失明[21]。因此,针对有近亲结婚等遗传背景新生儿,若出现不明原因脐带断端出血或皮肤淤斑、淤点,需提高警惕,尽快完善血浆Fg水平检测,早期进行Fg相关基因检测以明确出血病因,从而预防因创伤、手术等2次原因导致婴幼儿或成年期严重出血。
本研究文献检索的结果显示,关于FGG基因突变引起CA新生儿期发病的相关文献有限,纳入研究的仅有3例报道。新生儿期CA严重程度与突变的基因型有关,FGG基因纯合突变、复合杂合突变或移码突变,常发生先天性无Fg血症,患儿血浆Fg水平极低,临床症状重;而发生FGG基因错义突变时,由于患儿可合成Fg,保持其血浆Fg水平处于正常值的中等水平,因此CA患儿临床症状轻微并且异质性较高,临床诊断困难,更易被漏诊,这也在一定程度上解释了新生儿期起病的CA文献报道极少的原因。FGG基因错义突变的CA患儿,临床表现多为淤斑或脐带断端渗血,本例患儿即因为面部淤斑就诊的。此外,Fg γ二聚体(FGG二聚体)形成受损,可导致新生儿坏死性小肠结肠炎或脓毒症[22],但相关基因学研究较少。本例患儿及其母亲同样为FGG基因exon 8:c.1073C>A p.(Ser358Tyr)错义突变、杂合突变,患儿于新生儿期即出现面部淤斑症状,而其母亲无出血相关症状,因为母亲产前凝血功能检查结果显示异常,而被诊断为低纤维蛋白原血症。这提示,FGG基因杂合子错义突变也可有出血表现,但由于症状轻,常在成年期由于分娩或创伤后出血才被诊断。对于本研究检出的FGG基因错义突变,由于无相关文献报道及数据库收录,故笔者对该突变的遗传方式进行推断:由于母亲和患儿均有凝血功能障碍,并且患儿有面部淤斑临床表现,同时文献报道,FGG基因364和378位置的突变呈显性遗传,与本例突变位置358靠近,因此推断该突变呈显性遗传。总之,新生儿期该病的相关研究较少,需更多基因诊断信息扩充该病临床表型与基因型的相关性,为疾病预防提供遗传学资料。
Fg替代治疗是目前对CA患者采取的主要治疗方法,包括采取Fg浓缩物、新鲜冰冻血浆或冷沉淀静脉输注等治疗措施。对CA患者采取Fg治疗的目标水平,根据患者临床症状轻、重而不同,若患者合并颅内出血等严重出血或术前准备时,输注后血浆Fg水平需高于1.0 g/L,才可避免持续出血;而对轻微出血或非手术CA患者,则输注后0.5 g/L≤血浆Fg水平<1.0 g/L,即可避免出血症状,但迄今对于该类患者的Fg替代治疗剂量、间隔及持续时间,尚无统一标准[4]。文献报道,低Fg血症患者采取Fg替代治疗后,具有发生严重血栓疾病风险[23]。因此,Fg替代治疗同时,联合使用抗凝药物越来越被临床重视,但具体联合治疗方式迄今仍不统一。
综上所述,CA患儿新生儿期可以表现为脐带断端渗血、淤斑等,其疾病严重程度与基因型有关,但目前新生儿期被诊断罹患该病患儿的临床资料较少。采取单基因疾病高通量测序技术,可提高新生儿期CA诊断率,避免不必要的其他检查。此外,发现CA先证者Fg相关基因突变,不仅有助于优生优育,提供针对性强的遗传咨询信息,还能早期发现该基因突变携带者及预测CA相关并发症,避免该病患儿成年后不可控的大出血。遗传学诊断可进一步探讨CA发病机制,为本病的分子靶向治疗药物开发打下基础。对该病现有的主要治疗手段是在进行Fg替代治疗同时,需预防治疗相关血栓性疾病。
所有作者均声明不存在利益冲突

























