
患者,男,31岁,因"发现头痛、肋骨痛3个月余"入院。
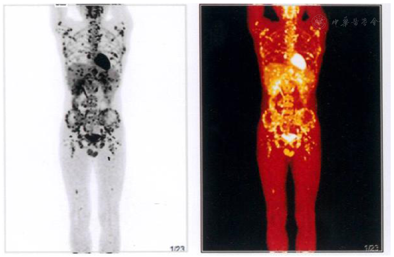
MRI检查发现颅内多发占位性病变。PET-CT检查示:1)左侧基底节、右侧顶叶及右侧鞍旁见高密度结节影,较大者约1.6cm×1.3cm。2)双侧甲状腺见多发低密度结节影,较大者约为0.6cm。3)右肺和胸膜见多发结节,较大者约1.5cm×1.0cm。4)腹腔、肝脏、脾脏、胃及前列腺内见多发结节,较大者约1.6cm×1.3cm。伴全身多发异常肿大淋巴结,及全身多发骨病灶。病灶放射性摄取均异常增高,SUV最大值为30.2。
病灶活检,活检组织行免疫组化、FISH基因检测及Sanger测序。
地塞米松+口服克唑替尼(赛可瑞) 250mg bid治疗。
随访时间16个月,局部病灶缩小消退,腹腔肿大淋巴结消退,患者病情平稳。
病理科;肿瘤内科;影像科;骨肿瘤科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
ALK阳性组织细胞增生症是Chan等[1]于2008年描述的一种罕见的疾病,累及实质性脏器及骨等多个部位,特征性的具有ALK阳性表达和/或ALK重排,尤其是KIF5B-ALK基因融合突变。该病变尚未归入WHO软组织肿瘤的分类中,国内尚未见报道,但其具有较特异的临床、免疫、分子改变和治疗靶点,为进一步探讨伴有KIF5B-ALK基因融合突变的ALK阳性组织细胞增生症的临床病理特点及鉴别诊疗要点,我们详细阐述一例多系统累及的病例,以期提高对该疾病的认识。
患者,男性,31岁,因"发现头痛、肋骨痛3个月余"入院。在外院MRI检查发现颅内多发占位性病变。随后PET-CT(图1)检查示:1)左侧基底节、右侧顶叶及右侧鞍旁见高密度结节影,较大者约1.6cm×1.3cm。2)双侧甲状腺见多发低密度结节影,较大者约为0.6cm。3)右肺和胸膜见多发结节,较大者约1.5cm×1.0cm。4)腹腔、肝脏、脾脏、胃及前列腺内见多发结节,较大者约1.6cm×1.3cm,伴全身多发异常肿大淋巴结,及多发骨病灶。病灶放射性摄取均异常增高,SUV最大值为30.2。临床诊断:倾向淋巴瘤。外院给予伽马刀及降颅压对症治疗,并行颅内立体定向穿刺术及胸膜病灶、右肺楔形切除术,未获明确诊断,遂于我院行骨穿刺活检术并进一步确诊治疗。患者无相关家族史、冶游史及手术史。
活检病灶灰红色质软。镜下见成片组织细胞增生,核膜不规则呈折叠状,见核沟,没有核仁,可见核分裂像,细胞无明显异型(图2,图3),见少量Touton巨细胞,间质出血伴散在淋巴细胞浸润。骨穿活检组织镜下见梭形的组织细胞均匀分布在髓腔中,部分细胞空泡状,胞质略嗜伊红色(图4)。
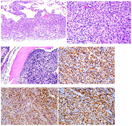
免疫组化:镜下组织细胞均ALK(5A4, 1:20; Thermo Fisher,Waltham, MA)细胞质/膜阳性(图5),CD163、CD68、PGM1均阳性表达(图6,图7),S-100和SMA部分阳性,AE1/AE3局灶阳性,肿瘤细胞Ki-67阳性指数约10%。CD21、CD23、CD1a、langerin、CD34、LCA、CD2、CD3、CD4、CD20、PAX-5均为阴性。
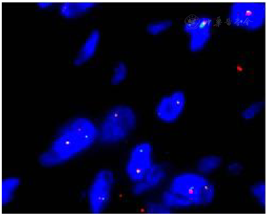
分子检测:ALK相关基因易位阳性(图8),Sanger测序结果示KIF5B-ALK融合突变。BRAF基因未见突变,EBER(-),B细胞基因重排(-)。
基于病变的受累范围、组织形态改变、免疫组化(组织细胞ALK阳性表达)和分子检测结果(KIF5B-ALK基因融合),该病例诊断为伴有KIF5B-ALK基因融合的ALK阳性组织细胞增生症。
需鉴别的其他病变包括:(1)Erdheim-Chester病:好发50-70岁男性,为系统性病变,常累及骨骼、心血管系统和腹膜后。X线特征性表现为双侧对称性斑块状或弥漫性髓腔硬化。镜下为泡沫状组织细胞、Touton巨细胞,表达CD68、CD163,不表达CDla、S-100、Langerin,>50%的病例显示BRAF-V600E突变[2]。而ALK阳性组织细胞增生症发病年龄更小,缺乏BRAF-V600E突变。(2)幼年性黄色肉芽肿:好发于婴幼儿头颈部的孤立性病变。表达CD68、CD163,不表达S-100、CD1a。免疫组化有助于鉴别。有些报道为播散性幼年性黄色肉芽肿的病例很可能是ALK阳性组织细胞增生症[3,4]。(3)朗格汉斯细胞组织细胞增生症:好发于青少年,常溶骨性骨质破坏并累及软组织及实质性脏器。常见破骨细胞样巨细胞,间质内见大量嗜酸性粒细胞。免疫组化表达CDla、S-100、Langerin,不表达组织细胞标记。常见BRAF-V600E突变。(4)软组织Rosai-Dorfman病:病变也可累及皮下、实质性脏器及骨组织。常有特征性吞噬淋巴细胞、浆细胞的伸入现象。免疫组化除CD163、PGM1组织细胞表达外还表达S-100,形态和免疫可鉴别。(5)上皮样纤维组织细胞瘤:成年男性多见的一种局限性皮肤肿瘤,大多数病例具有ALK重排和ALK过度表达。该肿瘤细胞呈上皮样,不表达S100和CD68,SQSTM1是最常见的ALK融合伴侣[5],而非KIF5B。此外还需与组织细胞肉瘤相鉴别,后者是一种侵袭性肿瘤,显示核多形性,易见核分裂像,ALK阴性表达可鉴别。
入院后给予地塞米松5mg静滴qd,并口服克唑替尼(赛可瑞)250mg bid治疗。一个月后继续予克唑替尼250mg、500mg qd交替口服。至今患者一直口服克唑替尼维持治疗。
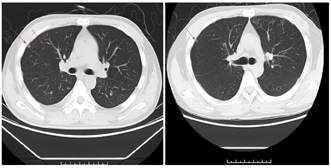
正规治疗前CT显示两肺多发小结节,大者约11mm,位于右肺中叶胸膜下。治疗1个月后,两肺只见少量小结节影,较大的约4mm(图9)。目前随访时间16个月,腹腔肿大淋巴结消退,双肺结节病灶明显减少缩小,病情平稳。
2008年Chan等[1]首次报道了3例ALK阳性组织细胞增生症,认为这是一种新的多发性系统性的组织细胞增生症。该组研究人员于2018年又增加了7例并总结报道[6],目前国外文献报道共约二十几例,国内尚无相关病例报道。总结这些病例,患者年龄谱从婴幼儿到成年人,部分患者表现为全身系统性病变,以肝脏、脾脏、肾脏、肺、结肠、皮肤、骨、软组织及中枢神经系统受累为主,部分患者表现为孤立性病变,累及鼻、声门下、皮肤、肝脏、乳腺和颅内海绵窦等部位,临床主要表现为肝脾肿大,贫血和血小板减少。男女患病比例相近,女性略多。我们报道的这例为男性患者,病灶累及颅内、双侧甲状腺、双肺、胸膜、腹腔、全身淋巴结及多处骨组织,其中甲状腺和胸膜为首次报道的累及部位。
形态学上,所有病变组织均表现为大片组织细胞浸润,细胞中等大小或稍大,胞质丰富、呈嗜酸性或空泡状。核呈卵圆形或梭形,染色质边集,核膜不规则呈折叠状,可见核沟,少数细胞质内含有吞噬的红细胞、色素或小空泡。可见数量多少不等的核分裂,无坏死。这些特征与其他组织细胞增生性病变类似,而不同的是,该病变多核巨细胞或Touton巨细胞比较少见,间质只见散在淋巴细胞浸润。病变内组织细胞均显示组织细胞标记阳性表达,如CD68、CD163、PGM1,部分表达S-100,其他标记均为阴性,也无BRAF-V600E突变。最为特异的是组织细胞ALK细胞质/膜阳性表达。
间变性淋巴瘤激酶(anaplastic lymphoma kinase, ALK)是一种受体型酪氨酸激酶,是肿瘤的一种驱动基因。随着分子检测技术的发展,我们认识到有一部分肿瘤与ALK基因易位相关,目前共报道了29个ALK伴侣基因[7]。但多数肿瘤通常都以某个独特的伙伴基因为主要类型,如ALK阳性间变性大细胞淋巴瘤的NPM-ALK,ALK阳性大B细胞淋巴瘤的CLTC-ALK[8],上皮样炎性肌纤维母细胞肉瘤中的RANBP2-ALK[9],肺腺癌的EML4-ALK[10]和上皮样纤维组织细胞瘤的SQSTM1-ALK[5]。ALK阳性组织细胞增生症以KIF5B为主要的伴侣基因,KIF5B-ALK基因融合仅在该肿瘤和少数ALK阳性非小细胞肺癌中检测到。ALK阳性组织细胞增生症其他的伴侣基因均为个例报告,如COL1A2-ALK基因融合[11],EML4-ALK基因融合[12],和TRIM33-ALK基因融合[13]。
ALK基因与不同的伙伴基因融合,导致ALK蛋白的着色定位模式也不同。如ALK阳性间变性大细胞淋巴瘤为NPM-ALK基因融合,ALK蛋白细胞质/核弥漫着色,ALK阳性大B细胞淋巴瘤为CLTC-ALK基因融合,ALK蛋白显示为细胞质颗粒性着色。我们这例为KIF5B-ALK基因融合,ALK蛋白显示为细胞质/膜着色。
对于伴有ALK融合基因阳性肺腺癌患者,应用ALK抑制剂后生存期延长[14],故ALK融合基因阳性被称为"钻石突变",肿瘤中含ALK重排的患者可从ALK酪氨酸激酶抑制剂治疗中获益。局限性病灶的ALK阳性组织细胞增生症患者,局部切除后一般预后良好。而表现为系统性病变的病例,包括不能手术的病人,临床症状比较严重,服用ALK抑制剂克唑替尼治疗后病情基本能得到控制,多数病人辅助化疗之下可逐渐恢复[6]。本例地塞米松及ALK抑制剂克唑替尼治疗16个月,病变也得到控制和缓解。
组织细胞增生性病变复杂多样,病理形态和免疫表型常有交叉。总结已报道的二十几例ALK阳性组织细胞增生症病例,发现其临床表现、细胞学形态、免疫表型及基因改变均较为一致,故我们认为ALK阳性组织细胞增生症可能是一种独特的临床病理实体。诊断主要是在组织细胞增生性病变的基础上表现出独特的免疫(组织细胞ALK阳性表达)及融合基因改变(KIF5B-ALK基因融合)。在组织细胞增生性病变中增加检测ALK的表达或易位,有助于识别该病变,ALK融合基因阳性为这类病变提供了靶向治疗的依据,ALK抑制剂的使用可能让该类病患得到更有效的治疗。
所有作者均声明本研究不存在利益冲突

























