
中年男性,因反复游走性关节痛就诊,却发现明显肝硬化表现,多次铜蓝蛋白水平均正常或接近正常,初期不考虑肝豆状核变性,故反复多次肝穿刺明确肝硬化病因,后发现其角膜K-F环显著阳性,转而再次行肝豆状核变性相关化验和基因筛查,最后明确本病后行驱铜及低铜膳食等相关治疗。
显著的角膜K-F环阳性,伴有肝硬化的临床表现,未见锥体外系表现及精神症状。
结合24小时尿铜,角膜K-F环,肝脏及头颅MRI检查,ATP7B基因筛查和家系共分离验证,临床和基因均明确为肝豆状核变性。
住院期间,予二巯丙磺酸钠注射液静脉排铜,出院后口服青霉胺序贯治疗,葡萄糖酸锌抑制铜吸收治疗,并嘱施行低铜膳食。
出院后,定期复查24小时尿铜,肝功能,肝脏B超,头颅MRI等检查,规律服用药物,病情渐进好转。
神经科;消化内科;风湿免疫科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
肝豆状核变性(Wilson’s disease, WD)是一种单基因遗传铜代谢障碍性疾病,其致病基因为ATP7B,基因突变后导致铜无法排出而沉积全身各脏器,最终使患者出现肝脏和神经系统等受累的复杂表现,若延误诊治,患者可因肝功能衰竭或严重的锥体外系表现而死亡[1]。然而,若早期明确诊断和给予规范治疗,患者生活质量和寿命可与常人无异,凸显早期诊断的必要性。我们报道1例因铜蓝蛋白正常而延误诊断的WD患者,旨在提高对本病临床异质性的认识水平和诊断效率。
男性,37岁,1年余前出现左肩部疼痛,无明显红肿,影响肩部上抬活动,持续3天左右自行好转,其后反复游走性关节疼痛,一般单发,腕部,手指,膝部,足部,肩部均有累及,持续数天可自行好转,发作间隔数周至数月,未予重视,7月前出现左侧食指疼痛伴肿胀,影响手部精细动作,遂就诊当地医院,住院期间完善相关检查,B超检查无意发现其有肝硬化表现,因担心病情,6月前转诊上级医院,查铜蓝蛋白为197mg/L(200-600),后再次复查为225 mg/L,因铜蓝蛋白水平几乎正常未首先考虑WD,予完善肝穿刺等相关检查考虑"自身免疫性肝炎",行免疫抑制剂相关治疗,后因无法耐受药物副作用,转诊于北京某两家肝病医院,再次肝穿刺检查,考虑患者为"重度小叶性肝炎"。2月前因手部关节再次疼痛,就诊当地医院,查体发现患者角膜K-F环阳性,临床拟诊WD,行ATP7B基因检测,提示患者携带R616W和R778L致病变异,为求进一步诊治,转至我院,住院期间完善铜蓝蛋白,24小时尿铜,ATP7B基因检测,头颅及肝脏MRI检查,类风湿因子,血沉等检查,患者否认家族史、冶游史及手术史,考虑患者有明确的肝病表现,角膜K-F环阳性,24尿铜增高,ATP7B基因和家系验证结果,临床和基因均可明确为WD,后给予患者静脉排铜治疗,出院后予口服青霉胺序贯治疗,并低铜膳食,嘱其定期复诊。
铜蓝蛋白水平检测:患者初始因关节痛在当地住院检查却发现肝硬化表现,排查病因时完善铜蓝蛋白检测,第一次铜蓝蛋白为197mg/L(200~600),第二次为225 mg/L(200~600),第三次为209 mg/L(200~600);
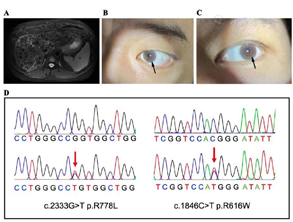
肝脏MRI结果:肝弥漫性病变,肝内多发再生结节考虑(图1A);
24小时尿铜:248.2μg /24h(15~60),静脉排铜治疗后复测:429.8 μg /24h(15~60);
角膜K-F环:双眼角膜周边可见棕黄色色素环(图1B,图1C);
ATP7B基因筛查结果:患者ATP7B基因明确为两个致病变异,分别是R778L和R616W(图1D),家系共分离提示其父亲为R778L携带者,母亲为R616W携带者。
根据《中国肝豆状核变性诊治指南2021》[2],对于原因不明的肝病表现,神经或精神症状,均应想到WD的可能。本例患者初期就诊过程中,也将WD列入鉴别诊断,但铜蓝蛋白基本正常,故未进一步排查其他临床指标,转向单纯肝硬化病因学筛查,辗转多家医院,反复多次行肝穿刺等有创检查,仍未明确诊断,最后因有经验的临床医生观察患者眼部,发现明显的角膜K-F环,方再次行WD相关筛查,最终明确WD诊断。
《中国肝豆状核变性诊治指南2021》指出[2],WD的诊断主要依据:1,神经和或精神症状;2,原因不明的肝脏损害;3,血清铜蓝蛋白降低和(或)24小时尿铜升高;4,角膜K-F环阳性;5,经家系共分离及ATP7B基因筛查明确患者2条染色体携带致病变异。若患者符合(1或2)+(3和4)或(1或2)+5均可诊断为肝豆状核变性。本患者肝脏MRI提示肝硬化,角膜K-F患者阳性,24小时尿铜大于100μg,ATP7B基因提示携带2个致病变异,父亲和母亲各携带其中一个致病变异,尽管血清铜蓝蛋白正常,仍可明确诊断为WD。
患者明确诊断为WD后,根据2021年指南建议,首先告知患者需长期维持低铜膳食;根据患者就诊时无明显神经精神症状,头颅MRI检查未见显著异常,所以给予患者二巯丙磺酸钠注射液1g静滴治疗6天;并予葡萄糖酸锌口服抑制铜吸收治疗;住院期间,青霉素皮试为阴性,嘱出院予口服青霉胺序贯治疗,逐渐增量,从31.25mg每次口服,逐渐增加至每天3次,每次125mg,并同时维生素B6片补充治疗。
根据患者静脉排铜后24小时尿铜结果为429.8 μg,建议其维持青霉胺,每次125mg,每天三次,餐前1小时口服治疗;继续服用1~3月,再次监测24小时尿铜,肝功能,血常规,肝脏B超,头颅MRI等,根据尿铜结果调整排铜药物治疗方案。
肝豆状核变性是一种遗传性铜代谢障碍性疾病,由于铜无法正常通过肝脏随胆汁排出体外,导致铜沉积在肝脏,后随血流波及全身,导致一系列复杂的临床症状。WD尽管是遗传性疾病,其确是一种可治疗的疾病,关键在早期诊断和规范化治疗,可使患者生存寿命与常人无异,凸显WD患者早期诊断的重要性。具有典型临床表现和生化指标的WD患者,根据2021年我国WD最新指南易被早期临床甄别[2]。但在实际临床工作中,仍有部分复杂型患者,偶因早期不典型的临床表现,铜蓝蛋白水平正常或角膜K-F环阴性,延误诊断,成为目前临床医师诊断过程中的棘手问题,提高临床医师对本部分诊断难点的认识水平,方能减少延误WD诊断的可能性。
ATP7B蛋白的核心功能一方面负责维持细胞内铜的稳态,另一方面促进铜和游离型铜蓝蛋白的结合,使其成为不易降解的结合型铜蓝蛋白[3]。因此,WD患者因为ATP7B基因突变,使其编码的ATP7B蛋白功能异常,导致WD患者表现为低铜蓝蛋白血症,也使其成为临床诊断中常用的经典指标。目前中国指南建议铜蓝蛋白低于120mg/L应引起临床医生高度重视,需行ATP7B基因检测,明确诊断[2]。然而,部分临床患者早期仅表现为轻微降低的铜蓝蛋白水平,甚至可为正常水平,干扰临床医师的正常诊疗,导致延误诊断。
前期发现WD患者在高雌激素血症的状态下,如怀孕期间和补充雌激素治疗,血清铜蓝蛋白水平可升高,甚至变为正常[4]。另一项研究指出,大约5%的WD纯合子患者,或高达50%的肝脏处于失代偿期的WD患者,也可表现为正常铜蓝蛋白水平,对这一发现的解释是,铜蓝蛋白是一种急性期反应产物,炎症期可促进其水平升高到正常范围[5]。另外,血清铜蓝蛋白的水平可以通过其具有铜依赖性氧化酶活性的酶促反应测量,或通过抗体依赖性测定,如放射免疫法、单向免疫扩散或比浊法。目前认为,这些方法的检测结果相互间是等效的[6],但临床上常规使用的免疫学检测法可能会高估铜蓝蛋白的水平,因其不能区分无铜结合的铜蓝蛋白和含铜的铜蓝蛋白[7]。
我们仔细分析本例患者反复就诊过程,尽管多次查血清铜蓝蛋白水平均为正常,但观察患者病史,初期就诊的主要诉求是全身游走性关节疼痛,就诊我院后查血沉偏高,提示体内处于炎症活动期,可能正因反复关节痛与WD并存,才导致患者铜蓝蛋白水平升至正常,造成阴性表现。尽管WD患者亦可表现为关节痛[8],但本患者反复游走性疼痛,可自行好转特点,血沉、类风湿因子异常,并请风湿科专科会诊,不考虑是WD所致。
尽管本患者铜蓝蛋白水平正常,倘若初期临床医师进一步行简单的角膜K-F环探查和24小时尿铜化验,也可避免本患者反复多次的就医和肝穿刺有创检查,以及延误诊断。临床医师需意识到,不管是铜蓝蛋白,角膜K-F环,以及24小时尿铜水平,均有假阴性可能,临床医师需正确把握临床诊断标准,意识到WD复杂的临床异质性,综合全面审视各临床诊断指标,避免WD的误诊漏诊。
所有作者均声明本研究不存在利益冲突

























