
因"体检发现左肾肿物1月余"入院。患者无腰痛、发热、血尿、尿痛、尿急等不适。既往双肾多发囊肿两年余,间断出现劳累后腰痛,规律复查。
生命体征平稳,心肺腹查体未见明显异常。专科查体双肾区无异常隆起,无叩击痛;双侧输尿管走行区无压痛,膀胱未充盈,耻骨上区无压痛。
术后病理提示为不能分型的肾细胞癌,考虑特殊类型肾癌。基因检测结果示FH基因突变。
行左肾根治性切除术。
术后恢复良好,无明显复发征象。
泌尿外科;病理科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
遗传性平滑肌瘤病相关肾细胞癌(hereditary leiomyomatosis and renal cell carcinoma, HLRCC)是由延胡索酸水合酶(fumarate hydratase, FH)基因突变导致的一种罕见的常染色体显性遗传病。患者易发生皮肤平滑肌瘤、多发性和早发性的子宫肌瘤以及肾癌或肾囊肿。肾肿瘤发病较早,主要表现为Ⅱ型乳头状肾细胞癌的特征,可为单个,双侧或多灶性病变[1]。我们报道一例以肾癌为主要表现的遗传性平滑肌瘤病,旨在提高对本病的认识,减少误诊和漏诊。
患者,男性,60岁。因"体检发现左肾肿物1月余"入院。患者自诉于1月前因双肾多发囊肿复查泌尿系CT增强提示左肾上极占位,双肾多发囊肿,双肾结石。进一步完善泌尿系MRI增强提示:左肾上极占位,伴左肾上段肾静脉内瘤栓。双肾多发囊肿。患者无腰痛、发热、血尿、尿痛、尿急等不适。现为进一步治疗入住我科。发病以来,患者精神欠佳,饮食尚可,睡眠可,两便正常,体重无明显变化。既往发现双肾多发囊肿2年,间断出现劳累后腰痛,规律复查,两年前于外院行肾囊肿穿刺术,穿刺抽液送病理提示未见肿瘤细胞。否认家族遗传病史及类似疾病史,个人史及婚育史无特殊。
体格检查:生命体征平稳,心肺腹查体未见明显异常。专科查体双肾区无异常隆起,无叩击痛;双侧输尿管走行区无压痛,膀胱未充盈,耻骨上区无压痛。
泌尿系CT增强(图1):左肾上部可见团块状稍低密度灶,边缘较清晰,大小约3.4cm,3.1cm,3.5cm,平扫及各期CT值约33HU,47HU,49HU,55HU。双肾实质内可见多个囊性低密度灶,边缘清晰,较大者位于左肾,直径约8.7cm,多期增强扫描病灶未见强化。双肾可见类圆形高密度灶,界清,较大者直径约1.1cm,CT值约74HU,增强扫描未见异常强化。影像学提示左肾上极占位。双肾多发囊肿(Bosniak Ⅰ-ⅡF)。双肾结石。肝多发囊肿。
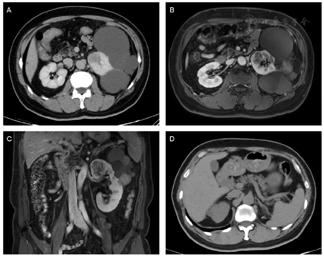
泌尿系MRI增强(图1):左肾可见多发大小不等囊性灶,双侧肾盏、肾盂、输尿管未见扩张,其内未见异常信号。左肾上部内侧可见结节状T1WI稍低T2WI稍高信号灶,DWI高信号,大小约3.3cm,3.1cm,4.3cm,反相位图像未见明确信号减低,病变延续进入左肾上段肾静脉分支,动态增强扫描可见不均匀轻至中度强化。双肾实质内可见多个囊性T2WI高信号,部分病变T1WI呈高信号及囊壁不均匀增厚,动态增强扫描未见异常强化,较大者位于左肾,直径约8.7cm。影像学提示左肾上极占位,伴左肾上段肾静脉内瘤栓。双肾多发囊肿(Bosniak Ⅰ-ⅡF),肝多发囊肿。
遗传性平滑肌瘤病的临床诊断的主要标准为:多处皮肤病变存在,其中至少一处组织学证实为平滑肌瘤。可疑标准为:(1)孤立性皮肤平滑肌瘤和HLRCC家族史;(2)Ⅱ型乳头状结构的早发性肾肿瘤;(3)在女性中,多发早发子宫平滑肌瘤。若FH基因存在胚系突变,HLRCC可明确诊断。
鉴别诊断:
是遗传性肾癌最常见的一种,往往表现为双侧多发的肾囊肿或肾癌,常合并中枢神经系统血管母细胞瘤、视网膜血管母细胞瘤、胰腺囊肿、肾上腺嗜铬细胞瘤、内耳淋巴囊肿瘤和附睾乳头状囊腺瘤等病变。VHL基因检测是VHL综合征诊断的金标准。
患者多在成年后出现双侧肾脏囊肿,随年龄增长,逐渐损害肾脏结构和功能。常常伴有PKD1或PKD2基因的突变。
是一种常染色体显性病。90%以上患者可以出现皮肤损害及神经系统病变。约80%患者存在肾脏囊肿或肾脏血管平滑肌脂肪瘤。基因检测发现TSC1或TSC2基因存在致病性突变可确诊为结节性硬化综合征。
患者初步诊断为左肾肿瘤并左肾静脉瘤栓,双肾囊肿。行"后腹腔镜左肾根治性切除术",切除左侧肾脏及脂肪囊。剖开左肾标本,肿瘤位于肾中上部内侧,无明显突出,直径约4cm,实性,切面白色,可见卫星灶,肾静脉内可见完整瘤栓,送病理检查。
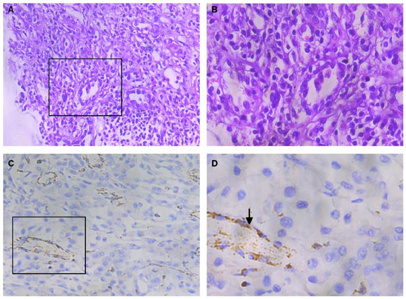
病理示:肾细胞癌,免疫组化不能完全确定组织分型(图2A,图2B,图2C,图2D)。肿瘤分级G2,部分G3,肿瘤大小4.5cm×3cm×3cm,浸润肾周脂肪,肾静脉内可见瘤栓,PT3a。手术断端未见肿瘤,肾脏多发单纯性囊肿,直径0.5~3.5cm。免疫组化:Vim(++),AE1/AE3(++),CD10(+),CA-9(-),CK(-),CD117(-),E-Cad(+++),FH(-),M630(+),TFE-3(+),HMB45(-),ALK(-)。
基因检测:取患者血液及肿瘤及癌旁正常组织送二代基因测序(Next Generation Sequencing, NGS),结果显示患者FH基因出现胚系杂合性突变,突变类型为截断突变,exon10, c.1499G>A, p.W500*,患者确诊为遗传性平滑肌瘤病。
患者术后恢复良好,伤口正常愈合。术后三个月后复查腹部CT平扫示(图1D):左肾切除术后,局部未见明显复发征象。建议患者定期监测,每年行腹部磁共振成像检查。
HLRCC是一种罕见的遗传性肾癌,由于对该病的认识不足,该病的发病率可能被低估[2]。研究报道,672名HLRCC患者中,474(71.5%)名患者出现皮肤平滑肌瘤,356(83%)名女性患者出现子宫平滑肌瘤,189(34.9%)名患者出现肾癌。平均诊断年龄为分别为28岁、32岁和36岁[3]。因为患有皮肤平滑肌瘤患者仅有微小的皮肤病变或无临床症状,很容易被患者忽略。尤其在男性患者中,患者可能仅表现为肾脏部位病变,给该病的诊治带来一定的困难。
HLRCC相关肾细胞癌主要为Ⅱ型乳头状肾癌。组织学标志性特征为肿瘤细胞内嗜酸性的大核仁及核仁外周空区,该特征常不均匀地出现在整个病变中,且目前报道的FH-RCC组织学图谱广泛,需要与其他肿瘤鉴别。与HLRCC诊断高度相关的两种免疫组化生物标记物分别是延胡索酸水合酶(FH)和S-(2-琥珀酰)-半胱氨酸(2SC)[4,5]。FH基因突变导致肿瘤细胞内的延胡索酸水合酶表达缺失,在肿瘤组织中染色往往呈阴性表达。FH蛋白缺失生成的延胡索酸往往导致细胞蛋白质水平的高琥珀酸化,可用2SC抗体检测检测该种化学修饰。肿瘤细胞的细胞质和细胞核通常表现出2SC阳性染色,在正常肾实质中染色呈阴性。对于FH-/2SC+的肾癌或高度怀疑HLRCC的患者,FH基因的胚系突变仍是诊断HLRCC的金标准。
HLRCC相关肾细胞癌大多数为高度侵袭性,预后较差[6]。与希佩尔-林道综合征及多发性结节硬化等遗传性肾癌不同,HLRCC肾癌患者即使原发肿瘤很小也可能发生远处转移,研究表明有肾癌表现的一半HLRCC患者出现肿瘤转移[3]。因此不推荐对HLRCC患者的肾肿瘤采用主动监测的方式,推荐对患者行保留肾单位手术或根治性肾切除术。目前,针对FH突变引起的转移性HLRCC患者暂无标准系统疗法。在HLRCC中,缺氧诱导因子HIF累积导致多种恶性肿瘤相关的信号通路发生变化,FH失活造成VHL非依赖性的HIF上升,同时导致NRF2激活[7]。因此,靶向下游通路的关键成分可抑制肿瘤的发生发展,如血管内皮生长因子VEGF,转化生长因子-α,表皮生长因子受体EGFR等。目前,针对VEGF和EGFR为靶点开展的贝伐珠单抗和厄洛替尼联合治疗晚期HLRCC相关肾癌的2期临床试验正在进行(AVATAR trail)[8],20年更新结果显示,患者的总客观生存率为64%(17/42),中位无进展生存时间为21.1个月。因此,NCCN临床指南推荐HLRCC患者使用贝伐珠单抗和厄洛替尼联合治疗。
HLRCC患者也可从免疫抑制剂中获益,目前关于PD-1抑制剂信迪利单抗(sintilimab)治疗HLRCC患者的临床试验正在进行(NCT04146831和NCT04387500)[9]。在国内FH突变相关肾癌患者的研究中,缓解持续时间在靶免联合及单独使用免疫治疗组的患者中客观缓解持续时间更久,即靶免联合相对于单独使用TKI药物对于FH突变患者能取得更好的治疗效果[10]。
所有作者均声明本研究不存在利益冲突

























