患儿,女,2017年10月份剖宫产,出生体重3.5千克,否认黄疸史,否认高热惊厥史。新生儿期肌张力高,偶有类似癫痫症状,产后4月不追物,现4岁,语言发育迟缓,运动能力落后。
面部、手部及足部皮肤正常,先天性玻璃体视网膜病变,颅脑核磁共振成像提示头颅稍小和胼胝体薄,视频脑电图监测显示为异常脑电图。
对先证者外周血DNA进行全外显子组测序,通过生物信息学的方法筛查可能的致病变异位点;利用Sanger测序对患儿及其父母进行突变位点的验证,分析家族遗传史;根据美国医学遗传学和基因组学学会指南对变异位点的致病性进行评估与分类。
在患儿的DOCK6基因中检测到c.143_146del和c.3190_3191del复合杂合变异,导致DOCK6蛋白翻译的移码和提前终止,产生p.T48Kfs*13和p.L1064Vfs*60突变蛋白,诊断患儿为2型Adams-Oliver综合征的临床罕见病例。后为夫妇双方提供遗传咨询,告知其可通过胚胎植入前遗传学检测或者产前诊断获得健康的子代。
夫妇双方因再生育需求来院就诊,因经济因素选择自然妊娠与产前诊断相结合的方式再次生育,患儿目前在其他医院进行相关症状的治疗。
遗传科;眼科;神经科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
Adams-Oliver综合征(Adams-Oliver syndrome, AOS)是一种罕见的发育性疾病,主要表现为先天性皮肤发育不全和末端横断肢体缺损(如截肢、并指、短指或少指),在活产儿中的发生率约为0.44/100000-1/10000[1,2]。AOS临床表现具有高度的异质性,可伴有其他器官尤其心血管系统和中枢神经系统的异常等[3,4]。根据致病基因和遗传规律的不同,AOS被划分为6个亚型,其中AOS1、3、5、6亚型为常染色体显性遗传,AOS2、4亚型为常染色体隐性遗传[5]。2型AOS的致病基因为细胞分裂因子6 (dedicator of cytokinesis, DOCK6),位于19号染色体p13.2区域,编码的DOCK6蛋白为一种非典型的鸟嘌呤核苷酸交换因子,在细胞骨架重塑和神经突起生长中发挥作用[6,7] 。本文中我们报道1例因DOCK6基因复合杂合突变导致的2型Adams-Oliver综合征,旨在提高对本病的认识,减少误诊和漏诊。
患儿,女,2017年10月份足月剖宫产,出生体重3.5 kg,第1胎第1产。面部、手部及足部皮肤正常,诉听力正常,新生儿期肌张力高,偶有类似癫痫症状。否认黄疸史,否认高热惊厥史。产后4月不追物,2019年1月经上海交通大学医学院附属新华医院诊断为渗出性玻璃体视网膜病变。语言发育迟缓,现4岁,会简单"爸爸妈妈"等单词。运动能力落后,坐不稳,不会爬行,不会走路。
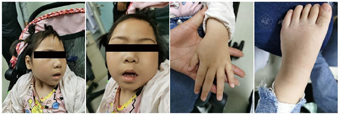
2018年3月颅脑核磁共振成像平扫提示中枢神经系统异常:头颅稍小,双侧侧脑室不规则扩大,呈波浪状,双侧侧脑室后脚变方,双侧侧脑室旁白质区明显减小;余脑内未见异常信号灶,脑池、脑裂未见明显增宽、加深,中线结构居中;小脑、脑干形态、信号未见异常,胼胝体薄。扫描范围双眼球信号不均匀,见条状T2WI稍低信号。
2021年5月重庆医科大学附属儿童医院视频脑电图检测显示异常脑电图:1、发作间期:醒睡期频繁可见两侧顶枕、后颞区为著高幅棘/尖-慢波阵发性出现伴全脑扩散,睡眠期为著;清醒期背景枕区优势不明显伴Ɵ活动增多;2、发作期:8次孤立痉挛样发作(双手突然上举,头部向右偏转),同步EEG可见广泛性不规则高幅尖样慢波发放,其后可重叠快节律。同步肌电可见肌电爆发,持续0.5-2秒。
父母表型正常,否认近亲结婚史,因再生育需求来我院就诊。家系图如图2,I:1代表患儿父亲,I:2代表患儿母亲,夫妇双方无明显临床症状;Ⅱ:1代表患儿。
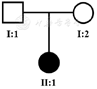
患儿父母签署了知情同意书,其个人信息经严格保密。本研究获得重庆市妇幼保健院伦理委员会批准。
患儿可能为常染色体隐性遗传病的受累个体,故对其进行全外显子组测序检测以及验证家族遗传史,具体方法如下:
采集患儿及其父母外周血2 ml,EDTA抗凝。利用QIAamp DNA Blood Mini试剂盒(德国Qiagen公司),按照说明书提取外周血DNA。采用Nanodrop 2000(美国Thermo fisher公司)检测DNA浓度并记录,然后放置于-20℃冰箱保存备用。
患儿DNA经超声破碎仪打断、末端修复、扩增、纯化等操作制备测序文库,采用特异性的捕获探针杂交富集目标区域的DNA序列。随后在Illumina NovaSeq 6000平台进行二代测序。使用NextGene V2.3.4软件将测序Reads与人的参考基因组(GRCh37/hg19)序列比对。单个核苷酸变异(SNV/indels)分析方法为:通过变异的人群频率数据库(dbSNP、ExAC、gnomAD等)对高频变异进行过滤,参考dbSNP、OMIM、HGMD、ClinVar等多种数据库对致病变异位点进行评估,应用SIFT、Polyphen2、MutationTaster、FATHMM等预测软件对变异的保守性和致病性进行预测。拷贝数变异(copy number variations, CNVs)分析方法为:对低质量的测序数据(编码序列平均覆盖度小于3×)进行剔除,通过均一化计算,分析出单/多个外显子的拷贝数变化,正常水平标注为1 (×2);缺失1个拷贝标注为0.5 (×1);缺失2个拷贝标注为0 (×0),即纯合/半合缺失。参考DGV、DECIPHER和OMIM等多种数据库和已发表文献对CNVs的致病性进行评估。根据美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics, ACMG)指南对SNV和CNV变异的致病性进行分类[8]。
针对DOCK6 c.143_146del和c.3190_3191del变异位点设计特异性扩增引物,对患儿及其父母目标序列进行Sanger测序验证。c.143_146del变异位于DOCK6基因第3号外显子,其特异性引物为Exon3_F: 5’-ATTGTCTCTTGCCTGGCCTC-3’; Exon3_R: 5’-TTGATGGGGTTGGCAGAGAC-3’。c.3190_3191del变异位于DOCK6基因第26号外显子,其特异性引物为Exon26_F: 5’-CAGCCTGGCTTTCTTCCTCA-3’; Exon26_R: 5’-GGCAAGAGAGGTGAAGGGTC-3’。热循环反应条件为:98℃ 2分钟,30个扩增循环(98℃ 10秒,55℃ 15秒,68℃ 30秒),68℃延伸5分钟。PCR扩增产物经胶回收纯化后进行Sanger测序,测序仪为ABI 3500。
利用分析软件Name Checker (https://mutalyzer.nl/name-checker)对突变蛋白进行命名,DOCK6 c.143_146del突变序列编码蛋白质为p.Thr48Lysfs*13,DOCK6 c.3190_3191del突变序列编码蛋白质为p.Leu1064Valfs*60。利用软件Illustrator for Biological Sequences (IBS, http://ibs.biocuckoo.org/online.php)对DOCK6基因和蛋白质进行模式图的绘制,并标明复合杂合变异的位置。
全外显子组测序结果显示,患儿19号染色体上DOCK6基因(NM_020812.4)存在3号外显子c.143_146del和26号外显子c.3190_3191del的复合杂合变异。PCR产物经Sanger测序,结果显示患儿父亲携带c.143_146del杂合变异,患儿母亲携带c.3190_3191del杂合变异,说明患儿的变异分别来自父亲和母亲(见图3)。
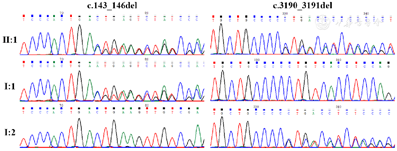
DOCK6基因位于19号染色体p13.2区域,含有48个外显子,主要编码含有2047个氨基酸的DOCK6蛋白(见图4)。DOCK6蛋白存在两个DOCK同源区域(Dock homology region,DHR),即DOCK同源区1(DHR-1),第548到714位氨基酸,能够结合磷脂[9];DOCK同源区2(DHR2),第1587到2023位个氨基酸,具有鸟嘌呤核苷酸交换活性[6]。c.143_146del变异和c.3190_3191del变异均会导致DOCK6蛋白翻译的移码和提前终止,分别产生p.T48Kfs*13和p.L1064Vfs*60突变蛋白,预测为丧失了正常功能的蛋白(见图4)。其中c.143_146del没有在相关临床病例中被报道过,c.3190_3191del在Adams-Oliver综合征的相关临床病例中被报道过[10,11]。
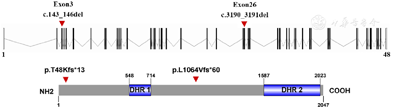
根据美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics, ACMG)提出的变异解读指南, c.143_146del变异为移码突变,导致DOCK6蛋白功能丧失,人群频率为0,位于DOCK6蛋白的重要功能区域,在不同物种间氨基酸序列高度保守,在反式位置上检测到致病变异,分类为可能致病(likely pathogenic)的变异;c.3190_3191del变异在Adams-Oliver综合征临床病例中以复合杂合状态被报道过,移码突变引起DOCK6蛋白功能丧失,分类为致病(pathogenic)变异。根据人类孟德尔遗传数据库(OMIM, Online Mendelian Inheritance in Man,https://omim.org/),该病例为DOCK6基因复合杂合突变引起的常染色体隐性遗传的2型Adams-Oliver综合征患儿。
夫妇双方因再生育需求来我院就诊,告知其可通过胚胎植入前遗传学检测(preimplantation genetic testing, PGT)即在胚胎移植前筛查胚胎染色体以排除携带遗传疾病及染色体异常的胚胎从而改善妊娠结局[12],或者产前诊断的方式获得健康的子代。夫妇双方因经济因素选择了自然妊娠与产前诊断相结合的方式进行再生育,于2022年1月份至我院产前诊断科室行羊膜腔穿刺和相关的基因检测。
在本文中,我们在一例以先天性玻璃体视网膜病变和中枢神经系统异常为主要表现的患儿DOCK6基因中,鉴定出c.143_146del和c.3190_3191del复合杂合变异。根据ACMG提出的变异分类指南,两个变异位点均会导致DOCK6蛋白翻译的移码和提前终止从而产生功能丧失的截短蛋白(p.T48Kfs*13和p.L1064Vfs*60),被分类为致病或可能致病的变异。该患儿被诊断为DOCK6基因复合杂合突变导致的2型Adams-Oliver综合征临床罕见病例,为夫妇双方提供遗传咨询并指导其健康生育。
人类基因组中包含11个编码DOCK家族成员的基因,分别命名为DOCK1到DOCK11,编码的鸟嘌呤核苷酸交换因子能够催化Rho GTPase RAC1/CDC42中GDP向GTP的转换,进而调控细胞运动和侵袭等生物学过程[13,14]。本文中报道的DOCK6 p.T48Kfs*13突变蛋白仅含有N末端60个氨基酸,DHR1和DHR2结构域的缺失导致RAC1/CDC42蛋白GEF催化活性的丧失。DOCK6 p.L1064Vfs*60突变蛋白含有1123个氨基酸,保留N末端序列的同时缺少含有DHR2结构域的C末端983个氨基酸,也导致了蛋白功能的障碍。另外自然界中存在一种无义介导的mRNA降解途径(nonsense-mediated mRNA decay, NMD),即当某个基因发生无义突变时,其转录出来的含有翻译提前终止密码子的mRNA会被NMD降解途径识别并带到细胞核外进行降解,从而防止对机体有害的截短蛋白的产生[15,16]。本文中患儿携带的DOCK6 c.143_146del和c.3190_3191del复合杂合变异均引起了DOCK6蛋白翻译的移码和提前终止,可能通过激发NMD信号通路引起突变mRNA水平的降低。DOCK6蛋白功能的丧失最终会导致Rho GTPase RAC1/CDC42蛋白活性的降低,进而引起血管生成和胚芽外胚层生长的障碍,最终导致四肢和颅骨血管受损[17]。
Adams-Oliver综合征以先天性皮肤发育不全和末端横断肢体缺损为特征,具有高度的表型异质性和遗传异质性,各临床症状的发生频率约为先天性皮肤发育不全(80%)、末端横断肢体缺损(85%)、心脏畸形(23%)、先天性毛细血管扩张性大理石样皮肤(20%)、中枢神经系统异常(常染色体隐性遗传个体30%)、眼科异常(<10%)、宫内生长受限(<10%)以及肾脏异常(<5%)[2,4,18],给临床诊断造成了极大困扰。DOCK6基因纯合或者复合杂合变异相关的2型Adams-Oliver综合征患者约占临床病例的17% [7]。据Varsome数据库(https://varsome.com/)记录,目前在DOCK6基因中共检测到263个变异位点,其中37个位点为致病变异(27个为致病变异,10个为疑似致病变异),55个变异位点的临床意义未明,171个变异位点为良性。37个致病变异分别包含14个移码突变(37.8%)、12个无义变异(32.4%)、7个剪接位点缺失变异(18.9%)、3个错义突变(8.1%)以及1个内含子突变(2.7%)。尽管中枢神经系统异常和视网膜病变在DOCK6基因突变相关的2型Adams-Oliver综合征患者中更为常见[4,18],目前尚无明确的基因型-表型调控机制被报道。
本文中报道的患儿主要临床表现为先天性玻璃体视网膜病变和中枢神经系统异常,未发现明显的先天性皮肤发育不全和末端横断肢体缺损,体现了Adams-Oliver综合征的临床表型异质性,有助于提高对本病的认识,减少误诊和漏诊。全外显子组测序对患儿的基因组DNA进行了遗传变异分析,检出DOCK6基因c.143_146del和c.3190_3191del复合杂合变异,均属于移码突变,扩展了DOCK6基因的突变谱;明确了患儿的致病原因,为家系的遗传咨询和产前诊断提供了理论依据。本案例补充了Adams-Oliver综合征的临床样本库,为进一步深入的基因型-表型调控机制研究提供了素材。
所有作者均声明本研究不存在利益冲突


























