
患儿,男性,6个月,以"反复抽搐1周"为主诉于2021年6月就诊于福建医科大学附属协和医院儿科。
患儿于5个月龄起病,主要表现为反复成串样痉挛发作及全面性发育迟缓,查体无特殊。
根据患儿临床表现及发作间期脑电图高度失律表现,诊断为West综合征。为明确病因完善影像学检查头颅磁共振成像提示无脑回及巨脑回畸形,送检外周血二代基因测序提示MACF1基因1号染色体39460714位点87号外显子上存在c.15266T>C(pMet5089Thr)的错义变异,其父母为野生型,符合常染色体显性遗传方式。正常人群数据库中未发现该变异。行蛋白质功能预测也提示该位点基因突变可能有害。
患儿入院后予ACTH联合托吡酯治疗2周后抽搐停止,后因呼吸道感染再次出现痉挛发作,遂调整抗癫痫药物为氨己烯酸、托吡酯。
患儿抽搐症状逐渐停止,随访至今,患儿未再出现癫痫发作,复查视频脑电图未见痫样放电。
儿科;神经科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
West综合征(West syndrome,WS)是多病因的发育性癫痫性脑病,发病高峰年龄为4~6个月[1],表现为痉挛发作、脑电图高度失律和发育迟滞[2,3,4]。越来越多的基因被证实与West综合征相关[3]。微管-肌动蛋白交联因子1 (microtubule actin crosslinking factor 1,MACFl)基因突变主要导致脑结构畸形,罕见与癫痫相关[5]。功能学研究表明MACF1突变可通过影响神经元迁移和分化导致大脑畸形、智力障碍、肌张力减退和癫痫发作[5,6,7]。本文总结1例携带MACF1突变的West综合征病例,经文献复习揭示MACF1基因与West综合征的联系。
患儿,男性,6个月,以"反复抽搐1周"为主诉于2021年6月就诊于福建医科大学附属协和医院儿科。患儿于5月龄首次出现抽搐,表现为双眼凝视,点头、拥抱样动作,成串出现,5-10余串/次,持续2~5 min后自行缓解,5~6次/d;发作间期神志清楚。无外院诊疗史。患儿系第1胎第1产,足月顺产,出生时无窒息、抢救史。患儿2个月能逗笑,3个月能抬头,但入院时不能逗笑,抬头不稳,不能翻身、独坐。父母体健,非近亲结婚,否认家族遗传病及传染病史。体格检查:体重8.5 kg(P50)、身长66.0 cm(P25)、头围42.0cm(P25)。外观无畸形,头发分布正常,无特殊面容,自主体位,四肢肌力、肌张力正常,膝腱反射可引出,病理征阴性。
实验室检查提示血常规、血生化、血氨、血乳酸等检查均无异常。视频脑电图(图1)提示:背景弥漫性慢波增多,全脑区可见大量高度失律。头颅磁共振成像MRI(图2):①巨脑回及无脑回畸形②双侧额顶叶硬膜下间隙增宽③双侧侧脑室增宽④透明隔间隙。Gesell发育量表测评:DQ 39.1。
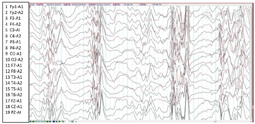
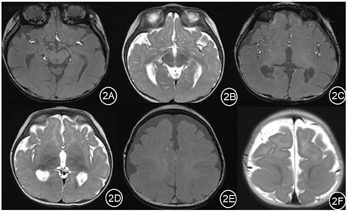
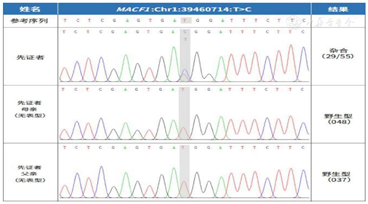
根据患儿临床表现及脑电图高峰失律图形,诊断为West综合征。为进一步明确病因,经福建医科大学附属协和医院医学伦理委员会批准(批准号:2020KY0108),患儿监护人签署知情同意书,采集患儿及其父母外周血行基因全外显子测序(whole exome sequencing,WES),并行Sanger测序进行验证。结果显示(图3)患儿染色体Chr1:39460714位点上MACF1基因存在新发突变(de novo),NM.012090.5:exon 87 c.15266T>C(pMet5089Thr),其父母该位点为野生型,经过Sanger测序验证证实,患儿为上述变异的错义突变,而其父母均未发现变异,符合常染色体显性遗传方式。正常人群数据库中未发现该变异。参照美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)评级为疑似致病性变异(likely pathogenic, LP)。
诊断:患儿系5月龄发病,表现为反复痉挛发作,伴精神发育迟滞,发作间期脑电图呈现高度失律,支持West综合征诊断。
鉴别诊断:(1)Lennox-Gastaut综合征:发病年龄通常在1-8岁,学龄前期为高峰,主要症状为智能减退、不典型失神及体轴性紧张性发作(可表现为仰头、点头或全身强直)。(2)良性肌阵挛性癫痫:多发生在6个月-3岁之间发育正常的儿童,表现为全身肌阵挛性抽搐,发作间期无异常表现。(3)早期肌阵挛性脑病:多于生后3个月以内发病,先为连续的肌阵挛,后发展为部分性的大量肌阵挛或强直性痉挛发作,脑电图为特征性暴发-抑制活动,可进展成高幅失节律,病情严重,可有精神发育停滞,第1年内即可死亡。
患儿入院后予ACTH联合托吡酯治疗,治疗2周后未再癫痫发作,复查脑电图未见明显高度失律。但患儿ACTH治疗的第26天,出现呼吸道感染致病情反复,再次出现痉挛发作,脑电图可见醒睡各期全脑区可见大量高度失律,遂停用ACTH,进入泼尼松序贯治疗,并加用另一抗婴儿痉挛症标准治疗药氨己烯酸,联合托吡酯共同使用。
加用氨己烯酸后患儿抽搐发作症状逐渐减停。经过5个月门诊及电话随访,患儿未再出现癫痫发作,期间于2021年9月复查视频脑电图未见痫样放电。
以"Epilepsy"、"West syndrome"、"lissencephaly"、"MACF1"、"癫痫"、"脑回畸形"、"婴儿痉挛症"为关键词分别检索PubMed、中国知网、万方等文献数据库,检索到相关文献21篇,相关文献阐述了MACF1参与大脑神经的发育过程,也提出MACF1是潜在的新型癫痫致病基因。但是目前国内外尚未有文献对MACF1所致West综合征进行单独报道,国外有两篇回顾性分析及一篇数据库分析提及共计12例癫痫患儿合并MACF1基因突变(不包括本例患儿),其中男性4例,女性8例,起病年龄3个月至5岁,包括本例在内的13例患儿均存在不同形式的癫痫发作、全面性发育迟缓、MACF1基因位点突变,其中10例合并脑回畸形,5例癫痫发作形式为痉挛发作。
MACF1基因是细胞骨架交联蛋白家族的一员,位于染色体chr1p34.3位点上,包含93个外显子,编码5430个氨基酸,MACF1包含三个主要结构域:一个由肌动蛋白结合域(actin-binding domain, ABD)和plakin结构域组成的N端结构域,一个由spectrin重复序列组成的棒状结构域,以及一个包含EF模体、GAS2相关蛋白(Gas2-related, GAR)结构域和甘氨酸丝氨酸精氨酸(Gly-Ser-Arg repeat, GSR)结构域的C端结构域[10]。检索Unitprot,提示MACF1是一种超过600kD的大蛋白,报道了常见的五种MACF1亚型,其中MACF1a广泛表达全身各组织器官,参与细胞骨架的构建、Wnt信号通路的正向调节、表皮细胞的迁移、囊泡转运及神经元的发育等过程。文献表明,MACF1的异常表达与脑回畸形的有重要的关系,并报告了MACF1异常表达所致脑回畸形相关West综合征的病例存在[5,6,9]。本报道患儿MACF1基因的新发错义突变(c.15266T>C)通过SIFT、Polyphen2、Mutation Taster等软件行蛋白质功能预测(图4),也提示该位点基因的突变可致蛋白质功能受到影响。

国内暂未见MACF1基因相关West综合征的报道。目前报道的与West综合征相关的致病基因虽罕见提及MACF1[3],但已有文献提示MACF1是潜在的新型癫痫致病基因。已有实验通过敲除MACF1构建小鼠纯合MACF1功能丧失(loss of function, LoF)突变体, MACF1完全缺失的小鼠胚胎的神经板始终未能形成原始条纹,并于胚胎期10.5-11.5天死亡。LoF突变体可通过破坏微管动力学、GSK信号及锥体神经元从而干扰神经元迁移,并可导致异常轴突延伸和引导[10]。大脑皮层发育过程与神经元的增殖、迁移密切相关[11],这证实了MACF1对于皮质发育及脑回形成起着重要作用。皮质发育异常越来越被认为是发育障碍和癫痫的原因[12]。无脑回畸形(Lissencephaly,LIS)或平滑大脑畸形和相关的畸形被称为皮质下带异位(subcortical band heterotopia,SBH),是与神经元迁移不足相关的典型畸形[13]。SBH的主要临床表现是智力低下和癫痫。癫痫几乎存在于所有患者中,并且在大约65%的病例中呈难治性。其中,超过90%的经典LIS患儿发生癫痫发作,约75%的病例在6个月前发病[14]。约35%-85%的典型LIS儿童发展成婴儿痉挛症[12]。综上所述,考虑本例患儿的癫痫发作病因与MACF1基因错义突变所致脑结构异常相关。
本例MACF1基因相关West综合征患儿应用ACTH联合托吡酯治疗效果欠佳,加予氨己烯酸治疗后患儿癫痫发作逐渐停止,复查脑电图高幅失律消失,提示氨己烯酸对MACF1基因突变所致West综合征可能有效,为该类患儿的治疗提供了一定的临床诊疗经验。
所有作者均声明本研究不存在利益冲突

























