患者,女,73岁。因"四肢麻木无力4个月,进行性加重2个月"入院。既往高血压、糖尿病、冠心病、高脂血症多年。
患者四肢麻木无力4个月余,进行性加重伴震颤、走路不稳2个月。查体:四肢近端肌力Ⅳ级,远端肌力Ⅴ-级。双上肢静止性及姿势性震颤。步基增宽、左右摇摆。双侧腕关节、膝关节以远针刺觉减退,双侧髋关节以远震动觉减退,关节位置觉正常。四肢腱反射未引出。
行肌电图、外周血血清学、脑脊液生化等检查,初步定位诊断为周围神经,定性诊断考虑为慢性免疫介导性周围神经病(CIN)。行腓肠神经切开活检术,术后病理及血清相关检查,提示为接触蛋白1(CNTN1)抗体相关的慢性炎症性脱髓鞘性多神经根神经病(CIDP)。
激素冲击序贯治疗,同时予缓解疼痛症状药物。
应用激素静脉冲击后患者麻木、震颤症状略减轻。激素序贯治疗后,患者症状再次反复,肢体无力、麻木症状较前无明显缓解。目前仍处于卧床状态,未恢复生活自理能力。
病理科;神经内科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
慢性炎症性脱髓鞘性多神经根神经病(chronic inflammatory demyelinating polyradiculoneuropathy,CIDP)是一类免疫介导的周围神经病,近年来,随着对部分CIDP患者血清中郎飞氏结/结旁区蛋白多个抗体的鉴定,人们发现有一些具有明确自身免疫抗体的患者常没有经典的CIDP病理学改变,却具有自己独特的分子及形态学特点,由于本病的异质性,不同病因的CIDP相对发病率较低,诊疗经验有限。本文介绍1例接触蛋白1(contactin-1, CNTN1)抗体相关的CIDP,具有CIDP的电生理特征,但临床表现有一定的特殊性,并且神经活检未见经典组织学形态,而活检组织平行神经长轴切面的超微结构特点、免疫组化组织化学染色、免疫荧光染色以及血清郎飞氏结相关抗体阳性,均支持CNTN1相关的CIDP诊断。因此,通过神经活检在组织学水平进行相关抗体检测,在不典型CIDP诊断中具有重要意义。
患者,女,73岁。主因"四肢麻木无力4个月,进行性加重2个月"入院。入院前4个月无明显诱因出现四肢麻木无力,麻木感远端明显,四肢轻度无力,尚不影响正常行走,伴双上肢及臀部间断针刺样疼痛,低头或弯腰时明显。2个月前舌头及口唇麻木、味觉下降,肢体麻木无力加重,右手抓握、持筷不稳,写字不能,伴右手抖动,下肢蹲起费力,步态不稳,走路时双下肢抖动,右下肢明显,臀部疼痛呈过电样,向下肢放射。1个月前面部麻木范围扩大,肢体无力加重,行走需拄拐。发病前否认感染、腹泻史。发病以来患者饮食、精神尚可,二便正常,时常失眠,体重无明显变化。
既往史:30年前车祸导致腰部软组织外伤,未骨折;冠心病、高脂血症20余年;高血压病史10余年;发现糖尿病2个月余,规律服药控制血糖。
个人史:生于北京市,职业农民。每年9月接种流感疫苗。无吸烟、饮酒史。否认家族性遗传病史。
入院体格检查:下肢轻度可凹性水肿。神清语利,双侧面部针刺觉对称减退,味觉减退,双侧掌颌反射(+)。四肢近端肌力Ⅳ级,远端肌力Ⅴ-级。双上肢静止性及姿势性震颤,右侧为著,双侧指鼻试验欠稳准。右侧跟膝胫试验欠稳准,Romberg试验睁、闭眼均不稳,步基增宽、左右摇摆。双侧腕关节、膝关节以远针刺觉减退,双侧髋关节以远震动觉减退,关节位置觉正常。四肢腱反射未引出。双病理征未引出。
其他辅助检查:血常规、血糖、肝肾功能、免疫检查、甲状腺功能、叶酸、维生素B12、同型半胱氨酸、血、尿本周蛋白未见明显异常。尿常规:尿蛋白3+;24 h尿蛋白定量2.28 g;抗M型磷脂酶2受体(PLA2R)抗体、肾小球基底膜抗体:均为阴性;抗神经节苷酯抗体(血清):GM1-IgG(-),GM1-IgM(+),GD1b、GQ1b IgG、IgM(-);脑脊液常规:细胞总数为2,白细胞数为0;脑脊液生化:蛋白179.4 mg/dl(12~60 mg/dl)↑,白蛋白841 mg/L(0~350 mg/L)↑,IgG 160 mg/L(10~40 mg/L)↑。脑脊液IgG合成率为19.7 mg/d(-9.9~3.3 mg/d);IgG寡克隆区带:(-);抗神经节苷酯抗体(脑脊液):GM1-IgG(-),GM1-IgM(-),GD1b、GQ1b IgG、IgM(-);抗神经抗原抗体(血清)CV2/CRMP5,PNMA2 (Ma2/Ta),Amphiphysin(-);抗Ri、Yo、Hu (-)。血清肿瘤标志物:CA242为30.22 kU/L(0~20 kU/L)↑;CA153为50.2 kU/L(0~25 kU/L)↑;CA 199为54.6 kU/L(0~39 kU/L)↑。颈椎MRI:颈4/5、5/6、6/7椎间盘突出;腰椎MRI:腰3/4、4/5、腰5/骶1椎间盘膨出;腰4椎体前滑脱Ⅰ度;腰骶丛神经根MR未见明显异常。PET-CT未见恶变征象。肌电图提示双侧运动神经CMAP、感觉神经SNAP、NCV下降,感觉神经损害重于运动神经,轴索损害伴脱髓鞘,右侧下降稍重于左侧;双侧F波、H反射未引出。震颤分析示负重时4~8 HZ高幅度震颤,右侧震颤较左侧明显。
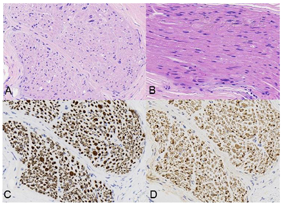
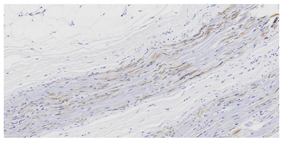
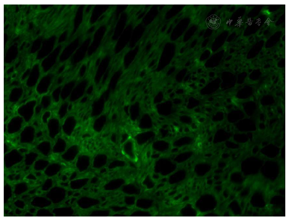
右侧腓肠神经活检病理显示神经束垂直切面,束内大直径有髓纤维轻~中度减少,残存有髓纤维可见轻度髓鞘松解及脱失,个别轴索肿胀变性,未见施万细胞增生或典型洋葱头样结构形成。见图1。电镜下神经束平行切面中郎飞氏结及结旁区可见点片状中等电子致密度物质沉积伴髓鞘终末处结构崩解或脱失。见图2。免疫组化染色:IgG4神经纤维纵切面及横切面中均可见阳性物质呈点灶状沉积于髓鞘,分布不均(图3),CD68(间质及束内极个别+),CD4(血管周及束内少数+),CD8(血管周及束内少数+),CD20(-)。免疫荧光染色示IgG髓鞘灶状阳性(图4),IgA、IgM及补体C1q、C3均为阴性。病理诊断: IgG4阳性的免疫性周围神经病,结合临床及血清学检查,考虑CNTN1抗体相关的CIDP。

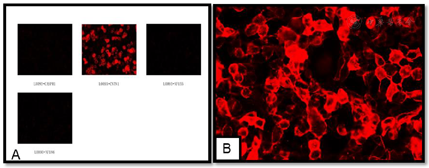
朗飞氏结相关抗体(脑脊液):CNTN1、CASPR1、NF155和NF186均为阴性;朗飞氏结相关抗体(血清):CNTN1阳性,CASPR1、NF155和NF186均为阴性(图5)。CNTN1的IgG分型检测:IgG4(1:100)。
定位诊断:面部针刺觉减退、味觉减退定位于三叉神经、面神经感觉纤维;意向性震颤、指鼻试验及步态欠稳准,定位小脑;四肢近端无力、腱反射消失、病理征阴性,下肢放射样疼痛,定位于周围运动神经,神经根受累为主;四肢感觉减退定位于周围感觉神经。
定性诊断:慢性进展性病程,主要表现为四肢近端无力合并末梢型深浅感觉障碍,血化验存在多项自身免疫相关指标异常,辅助检查示四肢感觉运动神经轴索损害和脱髓鞘,定性诊断考虑为慢性免疫介导性周围神经病(chronic immune-mediated neuropathies,CIN)。血清学郎飞氏结CNTN1抗体阳性,神经活检显示郎飞氏结/结旁区点片状中等电子致密度物质沉积伴髓鞘结构崩解或脱失,IgG4阳性,符合CNTN1抗体相关的CIDP。
鉴别诊断:(1)多灶性运动神经病:血清抗神经节苷脂GM1-IgM(+),但感觉神经受累更重,且肌电图无传导阻滞。(2)副肿瘤综合征:本例多个肿瘤标志物轻度升高且感觉神经手雷明显,需考虑此综合征累及周围神经系统,但本例也有运动神经受累表现,且没有明确肿瘤的证据。(3)血管炎性周围神经病:本例神经损害左右不对称,且自身抗体AHA阳性,需考虑,但神经活检病理结果不支持该诊断。
患者经济条件有限,拒绝使用美罗华治疗,同意使用激素冲击序贯治疗。予患者甲强龙240 mg冲击治疗,同时予加巴喷丁、依托考昔缓解疼痛症状。
应用激素静脉冲击后患者面部麻木、右上肢震颤症状较前减轻,可借助帮助缓慢行走。24 h尿蛋白较前下降。序贯口服激素治疗,患者症状再次反复,肢体无力、麻木症状较前无明显缓解。目前仍处于卧床状态,未恢复生活自理能力。
CIDP是一类由免疫介导的慢性运动感觉性周围神经病,临床异质性强。近年来,随着分子生物水平对郎飞氏结的深入研究,发现郎飞氏结及结旁区结构蛋白是CIDP患者免疫反应的特异性靶点,并在30%的CIDP患者血清中发现存在针对朗飞氏结/结旁区结构蛋白的自身抗体,包括神经束蛋白155(neurofascin 155, NF155)、CNTN1、接触蛋白相关蛋白1(contactin-1 associated protein1, Caspr1)和神经束蛋白140/186(neurofascin 140/186, NF140/186)[1,2,3,4]。这些抗体阳性的患者往往具有特定的临床特征和病理表现。
2013年Querol等[4,5]首次报道CNTN1是CIDP的生物标志物之一,并在46例CIDP患者中检测出3例CNTN1抗体阳性。CNTN1抗体阳性CIDP患者,男女比2.3:1;其临床特点:起病年龄偏大,平均发病年龄(63.0±13.5)岁,80%患者发病年龄超过60岁;多为亚急性起病,或慢性起病后迅速进展,进行性四肢无力,远近端受累相似,常有颅神经受累,有明显的感觉性共济失调,震颤以高波幅、意向性震颤为特点,常合并肾病综合征,对静脉丙种球蛋白治疗(IVIG)反应不佳,应用激素可能达到部分缓解,但仍可能复发;利妥昔单抗可能有长期、显著的疗效,且患者耐受性好[4,5,6,7,8,9,10,11,12]。本例患者为老年女性,慢性进展性病程,表现为感觉、运动均受累的周围神经损害,伴有震颤和共济失调,病程中有神经根刺激性症状,神经活检呈非经典型CIDP表现,血清郎飞氏结抗体示CNTN1阳性,最终诊断为CNTN1抗体相关的CIDP。本例患者临床与文献报道的CNTN1抗体相关的CIDP相符。但CNTN1抗体相关的CIDP还经常伴发膜性肾病,且通常抗PLA2R抗体阴性[7,8],本例虽尿蛋白升高,抗PLA2R抗体阴性,但未行肾活检,没有确诊膜性肾病依据,并且不除外高血压、糖尿病造成的肾损害。另外,本例患者在血清抗神经节苷酯抗体检测中发现GM1-IgM(+)。GM1-IgM阳性多见于多灶性运动神经病和Guillain-Barre综合征。也有个案报道GM1-IgM抗体阳性的CIDP[13],其临床以运动神经的非特异性损害特点,推测GM1-IgM抗体阳性CIDP可能是一种多灶性运动神经病和经典CIDP的中间型,可能进一步发展为远端获得性脱髓鞘性对称性神经病(DADS)。本例患者临床表现及肌电图均不符合GM1-IgM抗体阳性CIDP的特点,故不考虑该诊断。
在发病机制的研究中显示抗CNTN1-IgG4亚型抗体可穿透至结旁区,封闭CNTN1与其他结旁区蛋白的结合,引发髓鞘终末环松解,其病理表现为朗飞结区延长,结旁区结构破坏,髓鞘终末环从轴索上松解。而没有节段性脱髓鞘、施万细胞增生形成洋葱球样结构等经典型CIDP的病理改变[2,11,12,13,14]。本例患者腓肠神经活检可见大直径有髓纤维轻-中度减少,残存有髓纤维可见髓鞘松解及脱失,轴索肿胀变性,施万细胞轻度变性、增生,未见典型洋葱头样结构形成,结旁区可见点片状中等电子致密度物质沉积伴髓鞘结构崩解或脱失。免疫染色显示髓鞘内点状沉积IgG4免疫阳性物质。以上特点符合CNTN1抗体相关CIDP的病理特点,但很遗憾本病例未进行神经的CNTN1免疫染色,缺少直观的CNTN1抗体沉积的证据。
值得注意的是CNTN1作为细胞黏附蛋白在肿瘤的发生、转移起到重要作用,CNTN1分子的表达可作为乳腺癌、结肠癌等肿瘤预后的生物标记物。且有文献报道CIDP合并CNTN1抗体阳性患者发生肿瘤的病例。虽然该患者目前未发现恶性肿瘤的证据,但存在肿瘤标记物的升高,需密切监测肿瘤标记物情况,警惕潜在的恶性肿瘤。
所有作者均声明本研究不存在利益冲突


























