患儿,女性,3岁7月龄,因"TSH升高3年余"就诊,"先天性甲状腺功能减退"病史3年,在婴儿期出现了明显的生长发育落后和轻微的智力落后,期间使用L-T4替代治疗,反复调节药量效果不理想。
体重16.6 kg,身高102.5 cm,BMI 15.8 kg/m2。患儿无发热、无呼吸困难、无吞咽困难等不适,表现烦躁,心率快(>100次/min),体格检查颈部柔软,甲状腺区域未触及明显肿大。
患儿6日龄新生儿筛查TSH 193.81 mU/L (参考值<9 mU/L ),在当地妇幼保健院确诊为"先天性甲状腺功能减退"。5月龄时,因"不会翻身"就诊于当地医院儿童保健科,诊断为"发育落后"。10月龄17天,当地妇幼保健院评估:粗大运动差,精细运动中等偏下。我院基因检测结果显示患儿甲状腺过氧化物酶(thyroid peroxidase,TPO)基因复合杂合突变:外显子14,c.2395G>A(p.E799K)杂合突变,突变来源于母亲;外显子11 c.1949G>A(p.G650E)杂合突变,突变来源于父亲。确诊为"甲状腺分泌障碍2A型"。
患儿自出生后确诊先天性甲状腺功能减退后接受左甲状腺素钠片治疗,期间多次调整用量,目前用量为"12.5μg/d pod"、"50μg/d pod"交替。
截止末次随访,患儿3岁10月龄,体重16.5 kg,身高105 cm,较初诊时增长了2.5 cm,患儿生长发育好,甲状腺功能未见明显异常。
内分泌遗传代谢科;儿童保健科;耳鼻喉头颈外科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
甲状腺分泌障碍2A型(Thyroid dyshormonogenesis 2A,TDH2A,MIM#274500)是由甲状腺过氧化物酶(Thyroid peroxidase,TPO)基因纯合或复合杂合突变导致的一种先天性甲状腺功能减退症(congenital hypothyroidism,CH)。1992年,Abramowicz等[1]首次报道了TPO突变,其遵循常染色体隐性致病模式,造成甲状腺激素合成中碘的有机化缺陷。TPO基因突变的显著临床表现是永久性先天性甲状腺功能减退和甲状腺肿,根据缺陷的严重程度,甲状腺功能减退和甲状腺肿大的程度各不相同。本文报道了1例以生长发育落后为主要表现的甲状腺分泌障碍2A型,本患儿并没有文献中常报道的甲状腺肿的表现,旨在强调此病临床表型的多样性,提高对本病的认识,避免漏诊和误诊。
患儿,女,3岁7月龄,因"TSH升高3年余"就诊。患儿6日龄新生儿筛查TSH 193.81 mU/L (参考值<9 mU/L ),后在当地妇幼保健院确诊为"先天性甲状腺功能减退"(未见检查单),口服"左旋甲状腺素钠"治疗至今,期间多次调节药量(表1),现用量为"25μg/d pod"、"50μg/d pod"交替。以往甲状腺功能检查TSH明显升高,服药后常出现T4升高,或TSH升高,经常在TSH正常时,T4升高,反复调节药量,始终无好转。患儿近期化验FT4升高,FT3升高,TSH降低,为甲亢状态,表现为烦躁,心率快(>100次/min)。现为进一步诊疗遂来我院。自发病以来,患儿精神状态良好,体力情况良好,食欲及睡眠均良好,二便正常。
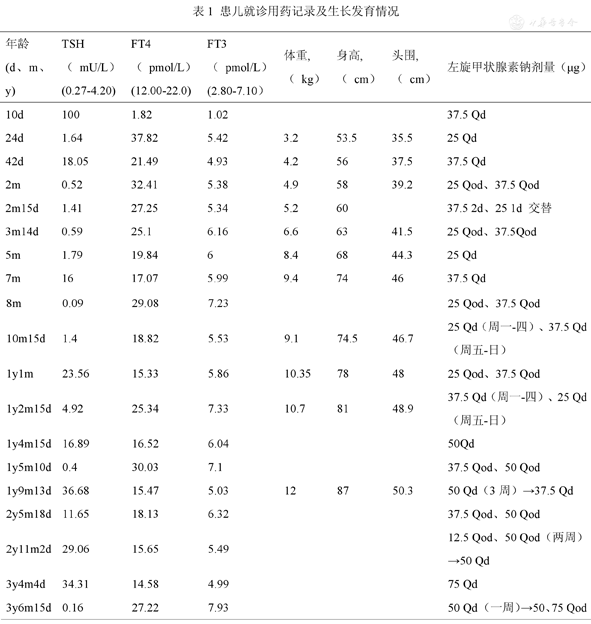
患儿就诊用药记录及生长发育情况
患儿就诊用药记录及生长发育情况
| 年龄(d、m、y) | TSH( mU/L)(0.27-4.20) | FT4( pmol/L)(12.00-22.0) | FT3( pmol/L)(2.80-7.10) | 体重,( kg) | 身高,( cm) | 头围,( cm) | 左旋甲状腺素钠剂量(μg) |
|---|---|---|---|---|---|---|---|
| 10d | 100 | 1.82 | 1.02 | 37.5 Qd | |||
| 24d | 1.64 | 37.82 | 5.42 | 3.2 | 53.5 | 35.5 | 25 Qd |
| 42d | 18.05 | 21.49 | 4.93 | 4.2 | 56 | 37.5 | 37.5 Qd |
| 2m | 0.52 | 32.41 | 5.38 | 4.9 | 58 | 39.2 | 25 Qod、37.5 Qod |
| 2m15d | 1.41 | 27.25 | 5.34 | 5.2 | 60 | 37.5 2d、25 1d交替 | |
| 3m14d | 0.59 | 25.1 | 6.16 | 6.6 | 63 | 41.5 | 25 Qod、37.5Qod |
| 5m | 1.79 | 19.84 | 6 | 8.4 | 68 | 44.3 | 25 Qd |
| 7m | 16 | 17.07 | 5.99 | 9.4 | 74 | 46 | 37.5 Qd |
| 8m | 0.09 | 29.08 | 7.23 | 25 Qod、37.5 Qod | |||
| 10m15d | 1.4 | 18.82 | 5.53 | 9.1 | 74.5 | 46.7 | 25 Qd(周一-四)、37.5 Qd(周五-日) |
| 1y1m | 23.56 | 15.33 | 5.86 | 10.35 | 78 | 48 | 25 Qod、37.5 Qod |
| 1y2m15d | 4.92 | 25.34 | 7.33 | 10.7 | 81 | 48.9 | 37.5 Qd(周一-四)、25 Qd(周五-日) |
| 1y4m15d | 16.89 | 16.52 | 6.04 | 50Qd | |||
| 1y5m10d | 0.4 | 30.03 | 7.1 | 37.5 Qod、50 Qod | |||
| 1y9m13d | 36.68 | 15.47 | 5.03 | 12 | 87 | 50.3 | 50 Qd(3周)→37.5 Qd |
| 2y5m18d | 11.65 | 18.13 | 6.32 | 37.5 Qod、50 Qod | |||
| 2y11m2d | 29.06 | 15.65 | 5.49 | 12.5 Qod、50 Qod(两周)→50 Qd | |||
| 3y4m4d | 34.31 | 14.58 | 4.99 | 75 Qd | |||
| 3y6m15d | 0.16 | 27.22 | 7.93 | 50 Qd (一周)→50、75 Qod |
患儿系G1P1,因迷信36周剖腹产,否认围生期缺氧窒息史,出生体重3.6 kg,身长不详。4月龄时,因"竖头不稳,不会翻身"就诊于"北京儿童医院",发育检查报告示:适应性轻度发育迟缓,大运动中度发育迟缓,精细运动轻度发育迟缓;语言正常;个人-社交中度发育障碍。5月龄22天时,因"不会翻身"就诊于当地医院儿童保健科,诊断为"发育落后",给予"PT(物理治疗)+OT(作业疗法)+多感官训练+推拿"治疗。10月龄17天,临河妇幼保健院评估:粗大运动差,精细运动中等偏下,继续上述康复治疗。患儿父亲身高183cm,母亲身高168cm,均健康,非近亲结婚,无类似疾病家族史。
停药一周后复查甲状腺功能五项,T3 1.74 ng/ml(参考值:0.7~2.5 ng/ml),T4 57 ug/L(参考值:40~170 ug/L),TSH 34.48 mIU/L(0.4~8 mIU/L), FT3 4.6 pmol/L(参考值:2.5~9 pmol/L),FT4 6.11 pmol/L(参考值:8.37~25.6pmol/L),抗体(-)。结果提示"高TSH血症"。
患儿因新生儿筛查而确诊先天性甲状腺功能障碍减退症,随即开始了口服左旋甲状腺钠(L-T4)的治疗,在婴儿期出现了明显的生长发育落后和轻微的智力落后,回顾病史可以发现患儿在婴儿期及幼儿期频繁地调整的L-T4的用量以达到甲状腺功能检测的正常,但是效果并不理想,并且本患儿不像常规的甲状腺功能减退症一样L-T4的需要量随着生长发育逐渐增加,我们考虑患儿是否存在特殊类型的甲状腺功能减退症导致的高TSH血症,如促甲状腺激素不敏感综合征,其特征为在无甲状腺肿的情况下,出现具有正常生物活性的高水平血清TSH,受影响患者的甲状腺正常或发育不良,血清TSH浓度高,血清T4和T3浓度正常或较低。我们在获得知情同意后采集患儿及其父母的血标本,提取外周血DNA,通过高通量测序仪进行基因检测(北京迈基诺医学检验研究所)。结果显示患儿甲状腺过氧化物酶(thyroid peroxidase,TPO)基因复合杂合突变:外显子14,c.2395G>A(p.E799K)杂合突变,突变来源于母亲;外显子11 c.1949G>A(p.G650E)杂合突变,突变来源于父亲。根据美国医学遗传学与基因组会学会(American College of Medical Genetics and Genomics, ACMG)指南分析,c.2395G>A为甲状腺分泌障碍2A型疑似致病性变异,c.1949G>A临床意义未明。
继予左甲状腺素钠片治疗,用量"25 μg/d pod"、"50 μg/d pod"交替,两周后调整药量为"12.5 μg/d pod"、"50 μg/d pod"交替。
目前正在继续治疗中,截止到最近一次随访,患儿3岁10月龄,体重16.5 kg,身高105 cm,较初诊时增长了2.5 cm。
人类甲状腺过氧化物酶(TPO)基因位于2p25染色体上,该染色体跨度>150 kb,其编码的甲状腺过氧化物酶是一种位于甲状腺细胞顶膜上的糖基化血红素蛋白,负责碘化物氧化、有机化和碘酪氨酸残基偶联,故TPO是甲状腺激素生物合成过程中的关键酶[2,3,4]。TPO缺陷是造成全组织碘缺陷(TIOD)的最常见原因,至今已有超过150种TPO基因缺陷被报道,在高加索人群中经常报道导致先天性甲状腺功能减退,但在东亚个体中发生的频率可能较低[5]。2015年一项针对中国广西CH患者进行TPO基因筛查的研究发现1%的CH是由TPO基因的致病性突变所致[4]。
在本例中,c.2395G>A和c.1949G>A分别导致p.E799K和p.G650E的氨基酸替换。p.E799K已有报道,该蛋白似乎正常插入细胞膜,突变的位置远离活性位点,但突变蛋白仍然没有酶活性,提示外显子14的酸性氨基酸与碱性氨基酸的交换可能导致分子的不正确折叠[6]。未见文献关于p.G650E突变的报道。曾报道G650E附近位点的R648Q突变,其功能也尚未得到验证,但所有携带R648Q突变的受试者均有严重的甲状腺功能减退,合理推测R648Q产生了重要的TPO活性缺陷。根据ACMG指南,G650E变异初步判定为疑似致病性变异(PM2+PM3(Trans)+PP3)。生物信息学蛋白功能综合性预测软件REVEL预测该突变结果为有害。然而,对mRNA的表达进一步分子研究是必要的,有助于更全面地了解这种突变对所得蛋白质结构和功能的确切影响。
本例患者表现为甲状腺功能减退,生长发育落后,高TSH血症,不伴甲状腺肿大,似乎并不符合TPO基因突变的典型表现,这表明TPO突变临床表型的多样性。TPO基因突变的显著临床表现是永久性先天性甲状腺功能减退和甲状腺肿,根据缺陷的严重程度,甲状腺功能减退和甲状腺肿大的程度各不相同[7],在文献中也有无甲状腺肿的报道[8,9]。TPO基因型和临床表型之间的关系尚未完全确立[4]。如果TPO酶活性完全丧失,患者可出现严重的永久性CH,表现为发育迟缓、TSH升高、T4降低;如果TPO酶的活性部分丧失,患者通常表现为轻度迟发性的甲状腺肿,伴有TSH升高或临界升高[10]。据报道,低于总TPO活性的15%,提示有甲状腺功能减退重度表型[11]。以往研究显示,60%~80%的患者甲状腺肿大,大多为多结节状,甲状腺肿大或可胸骨后侵犯而需手术干预。大约25%的儿童甲状腺结节被认为是甲状腺肿瘤的高危人群,而在成人中,这一比例为5%[12,13]。TPO基因缺陷的患者发生甲状腺癌之前文献报道过5例[14,15,16,17,18]。这部分患者甲状腺癌发病的一个可能解释是持续的高TSH刺激可促进甲状腺滤泡细胞的恶性转化[2,16,19],因此建议对甲状腺TPO基因异常患者进行仔细随访并及时进行遗传诊断[2,7]。
长期以来,严重的认知障碍一直被认为与延迟治疗或根本不治疗的CH患者的持续性疾病有关[2,20]。左旋甲状腺素是首选治疗方法,应尽早使FT4、TSH恢复正常[21]。先天性甲状腺功能减退的严重程度以及神经发育缺陷和TSH正常化的时间相关[22]。大多数早期治疗的CH患者观察到了正常的神经发育结果,但仔细监测认知、运动和行为发育是至关重要的,特别是在儿童时期,以确保发现任何需要早期干预的发育障碍,从而优化神经和社会发育结果[23]。在一些患者开始接受T4治疗后,甲状腺肿得以缓解。早期诊断和治疗可以改善远期预后[20]。而在TPO基因完全缺失的未治疗患者中,出现了导致智力低下和重度甲状腺肿的严重表型[24]。一些出生后立即接受治疗的患者发育正常,不出现甲状腺肿。本例患儿未见甲状腺肿,考虑是由于长期T4替代治疗,由于年龄过小,未出现相应表型也可能是原因之一,需要持续随访。另外,对于先天性甲减患儿进行康复训练(包括生活指导、亲子训练、物理疗法、感觉统合训练等)可提高其交际、理解能力和社会适应性,减轻残障的发生[25]。
综上,我们报告了一名中国患者的TPO基因中一个新的突变c.1949G>A和另一个c.2395G>A的复合杂合突变,拓展了TPO基因的突变谱,该患者表现为先天性甲状腺功能减退,高TSH血症,不伴甲状腺肿大。本报告强调了TPO突变临床表型的多样性以及及时有效的甲状腺素替代治疗对于提高此类患者生存质量的重要性。
所有作者均声明本研究不存在利益冲突


























