
患儿,男,1岁10月龄,因"间断抽搐发作1月余"就诊,首次发作年龄1岁9月龄,无明显诱因于睡眠时抽搐发作,表现为尖叫一声,然后双眼凝视、双手握拳、四肢抽搐,持续2~3 min缓解,抽搐后入睡,醒后精神正常,当时无发热、无呕吐、无腹泻等不适。起病后1月余再次类似发作2次。患儿为G2P2,足月顺产,否认出生窒息、抢救史,出生体重3.0 kg,8月龄抬头、18月龄独坐,20月龄能扶站,现不会独走,会发"baba mama"音。患儿母亲于11岁及20岁时分别抽搐1次,未就诊治疗,轻度精神发育迟缓;外婆多次抽搐发作,目前治疗不详,精神发育明显迟缓;家族成员否认近亲婚配,无其他类似疾病患者。
头围46.5cm,神志清,皮肤未见异常色素斑,无特殊面容,颅神经检查未见异常,双侧瞳孔等大等圆,对光反应灵敏,伸舌居中,颈软,心肺听诊未及异常,腹软,无压痛,肝脾肋下未触及,四肢活动自如,肌力肌张力正常,双侧膝腱反射可引出,病理征(-),无震颤,共济运动无异常。
患儿多次无热惊厥发作,Gesell评估示精神运动发育迟缓;头颅MRI脑实质未见异常信号,局部脑外间隙略宽;MRS示双侧基底节区及丘脑肌酸峰降低;高通量测序发现患儿X染色体SLC6A8基因剪接变异。结合患儿精神运动发育迟缓,癫痫发作,头颅MRS改变,故患儿诊断为SLC6A8基因变异导致的脑肌酸缺乏综合征1型。
丙戊酸钠口服抗癫痫发作治疗。
目前治疗半年余未再抽搐发作,但其他症状未见明显改善。
儿科;神经科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
肌酸在大脑、心脏和肌肉等高能量需求器官的能量代谢中起着至关重要的作用,促进二磷酸腺苷(ADP)通过磷酸肌酸合成三磷酸腺苷(ATP)[1]。肌酸在体内通过两个连续步骤进行生物合成,首先由精氨酸-甘氨酸脒基转移酶(AGAT)催化精氨酸和甘氨酸合成胍基乙酸和鸟氨酸,胍基乙酸再由胍基乙酸甲基转移酶(GAMT)催化转化为肌酸。肌酸通过钠和氯依赖性肌酸转运蛋白(CRTR)转运入细胞内[2]。脑肌酸缺乏综合征(cerebral creatine deficiencysyndrome,CCDS)是一组先天性肌酸代谢缺陷性疾病,共有3种类型,包括X连锁肌酸转运蛋白缺陷症(CCDS1)与常染色体隐性遗传肌酸生物合成缺陷,后者包括GAMT缺乏症(CCDS2)和AGAT缺乏症(CCDS3)。三种CCDS分别由SLC6A8、GAMT和GATM基因变异所致。CCDS常见临床特征为智力障碍、语言发育迟缓、自闭症样行为和癫痫[3]。该类疾病较罕见,国内仅有个案报道[4,5]。本文报告我院诊断的1例SLC6A8基因变异所致肌酸转运蛋白缺陷症患儿,探讨其临床特征与基因变异情况,旨在提高临床医师对该疾病的认识。
患儿,男,1岁10月龄,因"间断抽搐发作1月余"就诊,首次发作年龄1岁9月龄,无明显诱因于睡眠时抽搐发作,表现为尖叫一声,然后双眼凝视、双手握拳、四肢抽搐,持续2~3 min缓解,抽搐后入睡,醒后精神正常,当时无发热、无呕吐、无腹泻等不适。起病后1月余再次类似发作2次。患儿为G2P2,足月顺产,否认出生窒息、抢救史,出生体重3.0 kg,8月龄抬头、18月龄独坐,20月龄能扶站,现不会独走,会发"baba mama"音。患儿父亲体健,无抽搐史;母亲于11岁及20岁时分别抽搐1次,未就诊治疗,轻度精神发育迟缓;患儿有一姐姐4岁,生长发育同正常同龄儿,无抽搐史;外婆多次抽搐发作,目前治疗不详,精神发育明显迟缓;家族成员否认近亲婚配,无其他类似疾病患者。
体格检查:头围46.5cm,神志清,皮肤未见异常色素斑,无特殊面容,颅神经检查未见异常,双侧瞳孔等大等圆,对光反应灵敏,伸舌居中,颈软,心肺听诊未及异常,腹软,无压痛,肝脾肋下未触及,四肢活动自如,肌力肌张力正常,双侧膝腱反射可引出,病理征(-),无震颤,共济运动无异常。
Gesell评估适应性相当于11.2月龄,发育商51,中度发育迟缓;大运动相当于11.2月龄,发育商51,中度发育迟缓;精细动作相当于10.0月龄,发育商46,中度发育迟缓;语言相当于7.9月龄,发育商36,重度发育迟缓;个人-社交相当于9.1月龄,发育商42,中度发育迟缓。脑电图示背景活动5~6Hz θ节律,未见明显异常放电。头颅MRI脑实质未见异常信号,局部脑外间隙略宽;MRS示双侧基底节区及丘脑肌酸峰降低(图1)。
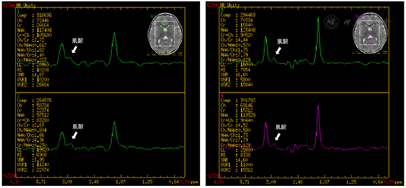
血肌酐14.4~20.0 umol/L(参考值11.0~34.0 umol/L),乳酸正常,甲状腺功能未见异常,血及尿串联质谱代谢筛查未见明显异常。
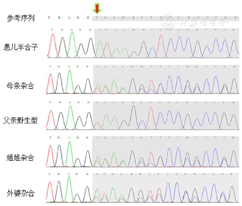
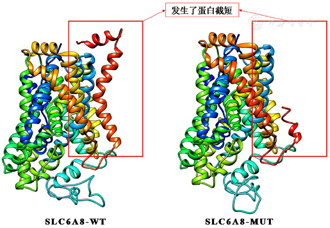
高通量测序及Sanger测序验证发现患儿X染色体SLC6A8基因剪接变异:NM_005629.4:exon12:c.1767+1_1767+2insA(染色体位置ChrX:153694890),患儿姐姐、母亲及外婆均为该变异杂合子(图2)。SLC6A8基因c.1767+1_1767+2insA变异尚未有文献报道,为经典剪接变异,第1767位核苷酸位于倒数第2个外显子上,通过SpliceAI在线预测发现,该基因12号外显子存在donor loss的同时,在上游99bp存在donor gain,所以推测有部分外显子被剪接。通过Mutalyzer在线预测该剪接变异的氨基酸序列,并通过SWISS-MODEL在线预测该基因野生型(WT)和突变体(MUT)的蛋白三维结构,该变异导致蛋白发生截短(图3)。根据ACMG指南,该变异为可能致病变异(PVS1_Strong+PM2_Supporting+PP4,该变异为经典剪接变异,第1767位核苷酸位于倒数第2个外显子上,故将PVS1降级为PVS1_Strong使用;PM2_Supporting:该变异在ExAC、gnomAD、千人基因组亚洲人群数据库中的发生频率极低或没有收录;PP4:患儿表型高度符合该基因变异所致疾病)。
患儿多次无热惊厥发作,Gesell评估示精神运动发育迟缓;头颅MRI脑实质未见异常信号,局部脑外间隙略宽;MRS示双侧基底节区及丘脑肌酸峰降低;高通量测序发现患儿X染色体SLC6A8基因剪接变异。结合患儿精神运动发育迟缓,癫痫发作,头颅MRS改变,故患儿诊断为SLC6A8基因变异导致的脑肌酸缺乏综合征1型。
该患儿予丙戊酸钠单药口服抗癫痫发作治疗,未添加肌酸、精氨酸及甘氨酸。
目前治疗半年余未再抽搐发作,但其他症状未见明显改善。
肌酸转运蛋白缺陷症(CTD)是一种X连锁疾病,是脑肌酸缺乏症(CCDS)的遗传性病因之一,由SLC6A8基因变异所致,该基因是溶质载体家族6的成员。该基因在大多数组织中表达,编码的肌酸转运蛋白是大脑和肌肉摄取肌酸所必需的转运体。Clark等[6]报道约1%不明原因男性智力低下与SLC6A8基因变异相关。
SLC6A8基因定位于Xq28,包含13个外显子,编码635个氨基酸,具有12个跨膜螺旋结构域(TM1-TM12),大部分错义变异和三碱基缺失发生在TM7和TM8结构域[7]。至目前为止,共报道140余种变异,主要为错义变异、无义变异,其次为小片段缺失、剪接变异。van de Kamp等[7]对SLC6A8基因不同变异与临床表型严重程度进行回顾性研究发现,错义变异可能致较轻的疾病表型,而片段缺失可能导致较严重的表型。本文报道1例SLC6A8基因c.1767+1_1767+2insA变异,为经典剪接变异,通过SpliceAI在线预测有部分外显子被剪接,该变异导致蛋白发生截短,根据ACMG遗传变异分类标准与指南,该变异为可能致病变异,且尚未有文献报道。
肌酸转运蛋白缺陷症由于SLC6A8基因变异致肌酸转运障碍,导致大脑、肌肉等能量需求高的器官发生功能异常。该病诊断年龄范围可从婴儿期至成年期,目前报道患儿预期寿命不受影响。临床主要表现为不同程度智力障碍、语言发育迟缓、自闭症样行为和癫痫。Salomons等[8]于2001年首次报道1例男性患儿表现为发育迟缓和肌张力减低,脑磁共振波谱未见肌酸峰;患儿成纤维细胞SLC6A8基因存在无义变异,导致肌酸摄取障碍。van de Kamp等对101例X连锁肌酸转运蛋白缺陷症患者进行回顾性研究,报道平均2岁独走,3.1岁会说单字,10岁以上患者中,14%没有语言表达,55%仅会说单词,31%仅会说单句;85%的4岁龄以下患儿存在轻至中度智力障碍,75%成年患者存在重度智力发育障碍;85%的患者存在行为问题,注意缺陷多动障碍和自闭症行为最常见;59%的患儿有惊厥发作,大部分患儿抗惊厥发作药物治疗可控制发作。除神经系统症状外,部分患儿还伴有消化道症状,包括新生儿喂养困难、呕吐、便秘、肝功能异常等[7]。部分患儿存在心血管系统异常,包括心肌病、室性期前收缩、长QT综合征等[9,10]。
X连锁肌酸转运蛋白缺陷症的女性携带者也可以有相关症状,但不如男性严重。Hahn等报道在男性表现为重度智力障碍、语言发育迟缓、行为异常和癫痫的家系中发现SLC6A8基因错义变异,女性携带者亲属表现为轻度智力低下和行为问题[11]。本例报道的患儿临床表现为发育迟缓、智力障碍、癫痫发作,家族中杂合携带该基因变异的母亲与外婆有惊厥发作史、精神发育迟缓,与既往报道的病例表现一致。
脑肌酸缺乏症患儿由于发育迟缓、行为异常,易被诊断为孤独症。YıldızY等报道一例男性孤独症患儿代谢筛查发现尿肌酸/肌酐比值增高,头颅MRS提示肌酸峰降低,进一步基因检测发现SLC6A8新发移码突变,最终确诊为肌酸转运蛋白缺陷症[12]。Battini等[13]报道一例经基因确诊的X连锁肌酸缺乏症男性患儿,在婴儿期表现为肌张力减低、喂养困难,3岁会独走和说单字,有热性惊厥发作,神经发育评估显示语言功能明显落后于非语言功能,因此脑肌酸缺乏症患儿的语言功能障碍更严重。临床有孤独症表现的患儿需进一步明确有无潜在病因。
由于患儿肌酸不能通过肌酸转运蛋白进而代谢为肌酐,因此患儿血及尿肌酐降低,尿肌酸与肌酐比值增高,此为一种可靠的筛查方法;杂合子女性该比值可在正常范围或轻度升高;本例患儿多次检测血肌酐正常低界。关于影像学检查,头颅MRI往往无明显异常或显示轻度异常。van de Kamp等[7]对肌酸转运蛋白缺陷症表型进行回顾性分析,76例进行头颅MRI检查者中53例存在轻度异常,包括轻度髓鞘形成延迟、胼胝体薄、轻度脑室或脑外间隙增宽及脑萎缩;66例进行头颅MRI检查者均显示肌酸完全缺失或严重降低。头颅MRS肌酸峰降低与基因检测有助于进一步确诊。Duran-Trio等通过敲入Slc6a8基因点突变建立CTD大鼠模型,发现这些雄性大鼠脑肌酸降低,尿肌酸/肌酐比值升高,并出现认知障碍和自闭症样行为[14]。因此,大鼠模型研究亦证实了SLC6A8基因突变者的临床表型。
CCDS的主要治疗方法为补充肌酸,AGAT缺乏症(CCDS3)患儿口服一水肌酸(300~400mg·d/kg)可显著改善发育,控制癫痫发作;GAMT缺乏症(CCDS2)除补充肌酸外(400~800mg·d/kg),还应补充高剂量L-鸟氨酸(400~800mg·d/kg)竞争性抑制AGAT活性和限制底物精氨酸的摄入以减少胍基乙酸(GAA)的合成,减轻GAA的脑损害;肌酸转运蛋白缺陷症(CCDS1)患儿肌酸内源性合成与转运均受影响,目前对于CCDS1的治疗疗效尚不确定[15]。Dunbar等[16]对肌酸转运蛋白缺陷症的治疗进行系统综述,28例患儿纳入分析,其中2例口服一水肌酸治疗,7例接受精氨酸治疗,2例口服一水肌酸和精氨酸,17例患儿口服一水肌酸、精氨酸和甘氨酸,28例患儿中有10例(36%)脑肌酸浓度增加和/或认知得到改善,且90%临床有改善的患儿均在9岁前接受治疗。Bruun等[17]提出肌酸、精氨酸和甘氨酸联合治疗可能阻止男性患儿疾病进展,并可改善女性患儿的临床症状。Schjelderup等[18]报告甜菜碱添加治疗成人肌酸转运蛋白缺陷患者耐受性好,患者语言和运动功能得到改善,因此甜菜碱可能成为治疗肌酸转运蛋白缺陷症的新疗法。El-Kasaby等[19]报道SLC6A8基因变异可导致转运蛋白错误折叠,这种突变体与内质网中钙连接蛋白结合,影响其在质膜发挥转运功能,同时发现4-苯基丁酸(4-PBA)与突变细胞共孵育可逆转转运蛋白突变体的内质网功能与质膜表达,提高转运体活性,为肌酸转运蛋白缺陷症的精准靶向治疗研究提供了依据。
肌酸转运蛋白缺陷为X连锁遗传性疾病,应向患儿家庭提供遗传咨询。男性患儿表型正常的父亲可正常生育;如果一个家庭生育有多个男性患儿,其母亲可能为杂合携带者;如果患儿为SLC6A8基因新生变异,尚不排除母亲体细胞或生殖细胞嵌合可能。携带者母亲每次怀孕都有50%的机会将致病变异遗传给下一代,遗传致病变异的男性将会发病,而遗传致病变异的女性将会杂合携带,但可能存在智力发育和行为问题。
因此,对于精神运动发育迟缓,尤其是语言发育障碍,伴癫痫发作、自闭症样异常行为的患儿,建议进行肌酸代谢筛查和头颅MRI检查,基因检测有助于进一步明确脑肌酸缺乏症的精准分型,指导治疗及改善预后。本研究报道了SLC6A8基因的新变异,丰富了该基因变异数据库。未来多中心的临床与转化研究将有助于进一步阐明脑肌酸缺乏症的自然病程、病理生理机制,及指导临床个体化精准治疗。
所有作者均声明本研究不存在利益冲突

























