
先证者,女性,20岁,因"腹泻伴体重下降1年"入院。1年前患者无明显诱因出现腹泻,每日3~5次,便稀或不成形,粪便中可见油滴,体重进行性下降约7kg,10个月前无明显诱因出现停经,未诊治。5 d前因腹泻次数较前增加,每日5~10次,无腹痛、黏液、脓血、里急后重等不适,无明显多饮,多尿等不适,就诊当地医院查空腹血糖大于20mmol/L(具体不详),为进一步就诊我院,门诊以"糖尿病"收入我科,病来精神饮食可,大便如前述,小便次数较前增多,具体不详。患者既往体健。第1胎第1产,足月顺产儿,母亲孕期无特殊疾病及用药史,自幼智力及生长发育正常无疫区长期旅居史,无吸烟、饮酒等不良嗜好、无毒物、放射性物质及职业粉尘接触史,无性病及冶游史
神志清楚,查体合作,面容正常,无特殊面容或黑棘皮病,全身浅表淋巴结未扪及肿大,心率95次/min,心律齐,心音正常,各瓣膜区未闻及杂音。双肺呼吸音清,未闻及干湿啰音及胸膜摩擦音。腹平,全腹柔软,无压痛及反跳痛,腹部未触及包块,肝、脾脏肋下未触及。双足底10g尼龙丝感觉减退,四肢痛、温、震动觉存在。
基因分析送北京迈基诺医学检验所,进行目标区域捕获测序:利用捕获芯片将目标区域的DNA片段进行富集后再借助高通量二代测序平台进行测序。本研究采用目标序列捕获芯片(MyGenostics, GenCap)对98个已知的垂体、肾上腺和胰腺相关候选基因进行捕获(表2)。捕获的具体过程如下:将基因组DNA随机打断成片段,与Illumina PE接头寡核苷酸混合物链接,对产物进行链接介导的聚合酶链式反应(ligation-mediated Polymerase chain reaction,LM-PCR)扩增纯化后得到DNA文库,并对其进行质量检测,将上述PCR产物与目标区域捕获芯片进行杂交以富集目标区域序列,借助Illumina HiSeq X ten测序平台对捕获的序列进行测序,并对原始数据进行初步处理,包括图像识别和样本区分。
给予生活方式干预,限制脂肪、蛋白质摄入,补充微量元素与维生素,外源性胰酶制剂及胰岛素治疗,入院时五点血糖19.8-18.0-31.8-23.4-17.7 mmol/L,予门冬胰岛素4U 3U 3U三餐前皮下注射,甘精胰岛素6U睡前皮下注射,患者血糖一直比较高,逐渐增加胰岛素剂量,1周后五点血糖为17.4-11.2-23.6-20.4-13.5mmol/L,胰岛素调整为门冬胰岛素20U 14U 9U三餐前皮下注射,甘精胰岛素20U睡前皮下注射,患者血糖逐渐好转,偶有低血糖发生,再治疗1周后五点血糖为7.6-5.9-12.3-9.6-8.8 mmol/L,胰岛素调整为门冬胰岛素2U 10U 9U三餐前皮下注射,甘精胰岛素12U睡前皮下注射,患者病情好转出院。
3个月后电话复诊患者仍予胰岛素治疗,偶有低血糖发生,胰岛素剂量调整为门冬胰岛素2U 6U 6U三餐前皮下注射,甘精胰岛素8U睡前皮下注射,空腹血糖7.0mmol/L左右。
内分泌科;消化内科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
遗传性胰腺炎(hereditary pancreatitis,HP)是一种罕见的胰腺疾病,Mayo临床中心1952年由comfort首次描述[1],其诊断标准包括胰腺疾病的症状、家族史和腹部平片上钙化表现,现将我科2018年收治一例女性糖尿病患者及其家族患者的诊治情况报道如下。
先证者,女性,20岁,因"腹泻伴体重下降1年"入院。1年前患者无明显诱因出现腹泻,每日3~5次,便稀或不成形,粪便中可见油滴,体重进行性下降约7kg,10个月前无明显诱因出现停经,未诊治。5天前因腹泻次数较前增加,每日5~10次,无腹痛、黏液、脓血、里急后重等不适,无明显多饮,多尿等不适,就诊当地医院查空腹血糖大于20mmol/L(具体不详),为进一步就诊我院,门诊以"糖尿病"收入我科,病来精神饮食可,大便如前述,小便次数较前增多,具体不详。
患者既往体健。第1胎第1产,足月顺产儿,母亲孕期无特殊疾病及用药史,自幼智力及生长发育正常无疫区长期旅居史,无吸烟、饮酒等不良嗜好、无毒物、放射性物质及职业粉尘接触史,无性病及冶游史。
 未婚未育。
未婚未育。
患者父母非近亲结婚,家中奶奶爷爷外公外婆已去世,死因不详,现有两代共7名成员,第一代中其爸爸(Ia),46岁,无糖尿病,胰腺无钙化,妈妈(Ib),40岁,本次检查发现患糖尿病,胰腺CT提示胰腺萎缩并钙化,慢性胰腺炎待排,第二代中先证者有三个妹妹及一个弟弟,其中第二个妹妹13岁(IIc),1年前在当地医院诊断"1型糖尿病并酮症酸中毒",本次就诊查胰腺CT同先症者,现胰岛素治疗。第一个妹妹(IIb)17岁,本次发现糖尿病,症状和胰腺CT较先症者轻,弟弟,10岁(IId),没有糖尿病,胰腺检查正常,第三个妹妹(IIe)8岁,没有糖尿病,胰腺检查提示胰腺萎缩并钙化,程度较先证者轻(图1,表1)
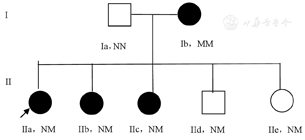
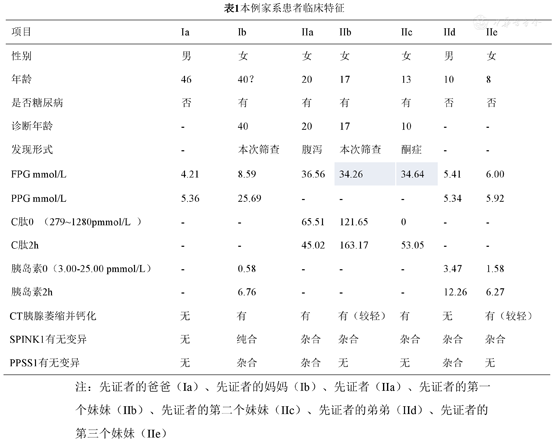
本例家系患者临床特征
本例家系患者临床特征
| 项目 | Ia | Ib | IIa | IIb | IIc | IId | IIe |
|---|---|---|---|---|---|---|---|
| 性别 | 男 | 女 | 女 | 女 | 女 | 男 | 女 |
| 年龄 | 46 | 40? | 20 | 17 | 13 | 10 | 8 |
| 是否糖尿病 | 否 | 有 | 有 | 有 | 有 | 否 | 否 |
| 诊断年龄 | - | 40 | 20 | 17 | 10 | - | - |
| 发现形式 | - | 本次筛查 | 腹泻 | 本次筛查 | 酮症 | - | - |
| FPG mmol/L | 4.21 | 8.59 | 36.56 | 34.26 | 34.64 | 5.41 | 6.00 |
| PPG mmol/L | 5.36 | 25.69 | - | - | - | 5.34 | 5.92 |
| C肽0 (279~1280pmmol/L ) | - | - | 65.51 | 121.65 | 0 | - | - |
| C肽2h | - | - | 45.02 | 163.17 | 53.05 | - | - |
| 胰岛素0(3.00-25.00 pmmol/L) | - | 0.58 | - | - | - | 3.47 | 1.58 |
| 胰岛素2h | - | 6.76 | - | - | - | 12.26 | 6.27 |
| CT胰腺萎缩并钙化 | 无 | 有 | 有 | 有(较轻) | 有 | 无 | 有(较轻) |
| SPINK1有无变异 | 无 | 纯合 | 杂合 | 杂合 | 杂合 | 杂合 | 杂合 |
| PPSS1有无变异 | 无 | 杂合 | 杂合 | 无 | 无 | 杂合 | 无 |
注:先证者的爸爸(Ia)、先证者的妈妈(Ib)、先证者(IIa)、先证者的第一个妹妹(IIb)、先证者的第二个妹妹(IIc)、先证者的弟弟(IId)、先证者的第三个妹妹(IIe)
血压100/86 mmHg,身高153 cm,体重37 kg,BMI15.80 kg/m2,神志清楚,查体合作,面容正常,无特殊面容或黑棘皮病,全身浅表淋巴结未扪及肿大,心率95次/min,心律齐,心音正常,各瓣膜区未闻及杂音。双肺呼吸音清,未闻及干湿啰音及胸膜摩擦音。腹平,全腹柔软,无压痛及反跳痛,腹部未触及包块,肝、脾脏肋下未触及。双足底10g尼龙丝感觉减退,四肢痛、温、震动觉存在。
1.实验室检查:
(1)常规检查:血常规、肝肾功能、胸片、心电图、电解质、血尿淀粉酶、大便常规、甲状腺功能、TGAb、TPOAb、PTH、ICA、GAD、IAA未见明显异常。
(2)血糖及糖尿病并发症筛查:尿糖4+,尿酮-;血糖36.56mmol/L,尿素2.29mmol/L,肌酐30.04μmol/L;糖化血红蛋白大于15%;C肽释放试验(表1),周围神经传导速度:左上肢神经末梢未见明显周围神经病变,右上肢及双下肢存在周围神经病变;尿白蛋白/肌酐112.01mg/dl;眼科会诊:右眼视力0.6、左眼视力0.04,晶体混浊,提示双眼白内障。
(3)性激素及皮质激素:LH:0.34U/L(卵泡期1.90~12.50),FSH:10.73U/L(卵泡期2.50~10.20U/L),PRL 110.82 mU/L(卵泡期59.00~619.00 mU/L),E2 51.78pmol/L(卵泡期71.60~529.20pmol/L),PRGE 1.25nmol/L(卵泡期0.48~4.45nmol/L),TSTO 1.27nmol/L(0.50~2.60nmol/L);ACTH (8:00) 19.10 pg/ml;皮质醇(8:00)24.60μg/dl(4.30~22.40μg/dl)。
(4)其他:CA199:36.75U/ml(0~27U/ml),CA242 :21.66 U/ml (0.00~ 20.00U/ml);IgG4 2.33g /L(0.03~2.01g /L);维生素D 5.98 ng/ml (30.00~70.00 ng/ml)。
(5)影像学检查:上腹部CT:提示胰腺外形缩小,密度不均,可见数个斑片状结节状高密度影,胰管呈不规则串珠状扩张,示胰腺萎缩、钙化(图2)。
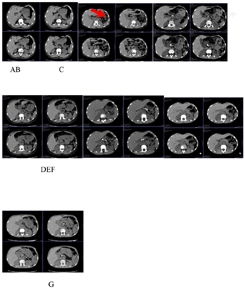
G为IIe)
(6)肠镜:回肠末端粗糙,凹凸不平,米粒样改变,考虑胰源性吸收不良,混合痔。
2.基因分析送北京迈基诺医学检验所,具体检测如下:(1)目标区域捕获测序:是利用捕获芯片将目标区域的DNA片段进行富集后再借助高通量二代测序平台进行测序。本研究采用目标序列捕获芯片(MyGenostics, GenCap)对98个已知的垂体、肾上腺和胰腺相关候选基因进行捕获(表2)。捕获的具体过程如下:将基因组DNA随机打断成片段,与Illumina PE接头寡核苷酸混合物链接,对产物进行链接介导的聚合酶链式反应(ligation-mediated Polymerase chain reaction,LM-PCR)扩增纯化后得到DNA文库,并对其进行质量检测,将上述PCR产物与目标区域捕获芯片进行杂交以富集目标区域序列,借助Illumina HiSeq X ten测序平台对捕获的序列进行测序,并对原始数据进行初步处理,包括图像识别和样本区分。
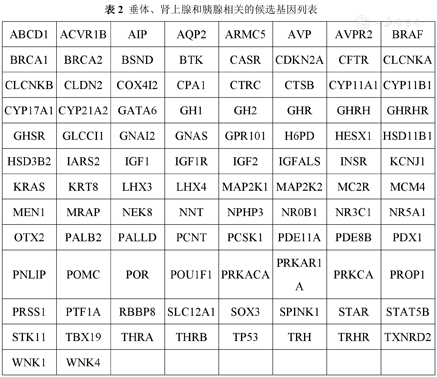
垂体、肾上腺和胰腺相关的候选基因列表
垂体、肾上腺和胰腺相关的候选基因列表
| ABCD1 | ACVR1B | AIP | AQP2 | ARMC5 | AVP | AVPR2 | BRAF |
| BRCA1 | BRCA2 | BSND | BTK | CASR | CDKN2A | CFTR | CLCNKA |
| CLCNKB | CLDN2 | COX4I2 | CPA1 | CTRC | CTSB | CYP11A1 | CYP11B1 |
| CYP17A1 | CYP21A2 | GATA6 | GH1 | GH2 | GHR | GHRH | GHRHR |
| GHSR | GLCCI1 | GNAI2 | GNAS | GPR101 | H6PD | HESX1 | HSD11B1 |
| HSD3B2 | IARS2 | IGF1 | IGF1R | IGF2 | IGFALS | INSR | KCNJ1 |
| KRAS | KRT8 | LHX3 | LHX4 | MAP2K1 | MAP2K2 | MC2R | MCM4 |
| MEN1 | MRAP | NEK8 | NNT | NPHP3 | NR0B1 | NR3C1 | NR5A1 |
| OTX2 | PALB2 | PALLD | PCNT | PCSK1 | PDE11A | PDE8B | PDX1 |
| PNLIP | POMC | POR | POU1F1 | PRKACA | PRKAR1A | PRKCA | PROP1 |
| PRSS1 | PTF1A | RBBP8 | SLC12A1 | SOX3 | SPINK1 | STAR | STAT5B |
| STK11 | TBX19 | THRA | THRB | TP53 | TRH | TRHR | TXNRD2 |
| WNK1 | WNK4 |
(2)生物信息分析:将原始测序数据去除污染和接头序列,然后利用BWA软件(http://bio-bwa.sourceforge.net/)将过滤后的序列比对到NCBI数据库人类基因组参考序列(hg19)上,利用GATK软件(https://software.broadinstitute. org/gatk/)分析得出单核苷酸变异(single nucleotide variation,SNV)和插入缺失突变(inserts and deletions,INDEL)的相关信息。然后通过ANNOVAR软件(http://annovar.openbioinformatics.org/en/ latest/)对所有的SNP和INDEL进行注释。筛选掉正常人数据库中频率小于0.05的突变位点,正常人数据库包括千人基因组计划(http://www.1000genomes.or/)、Exome Variant Server(http://evs.gs.washington.edu/EVS/)和EXAC(http://exac.broadinstitute.org/)。错义突变使用SIFT(http://sift.jcvi.org/),PolyPhen-2 (http://genetics.bwh. harvard.edu/pph2/),MutationTaster(http://www.mutationtaster.org/)和GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html)软件进行致病性预测和保守性预测,剪切位点的改变用SPIDEX(http://www.deepgenomics. com/spidex)软件分析其致病性。
(3)PCR扩增和Sanger测序:经过分析筛选得到的获选变异位点利用PCR和Sanger测序验证。PCR引物对利用Primer 3.0在线软件(http://primer3.ut.ee/)设计。PCR产物经过Sanger测序并在ABI 3130 Genetic Analyzer (Applied Biosystems)上进行分析。
(4)患者及家系DNA结果(表3):患者Ib的SPINK1基因5号外显子194+2T>C发生纯合突变,编码区第194+2号核苷酸由胸腺嘧啶变异为胞嘧啶,导致剪接突变,IIb、IIc、IId、IIe SPINK1基因194+2T>C发生杂合突变,Ib、IIa、IId患者PRSS1基因7号外显子发生杂合突变,编码区第623号核苷酸由鸟嘌呤变异为胞嘧啶,导致错义突变。
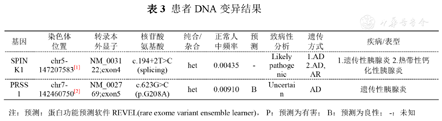
患者DNA变异结果
患者DNA变异结果
患者的临床特点是青年女性,发病年龄轻,脂肪泻,有糖尿病家族史,体形消瘦,无腹痛,无酗酒史,不耐受油腻饮食,无酮症倾向的糖尿病,胰岛功能较差,需要胰岛素控制血糖,CT发现胰腺萎缩并弥漫性钙化和基因检测符合遗传性胰腺炎的遗传规律。根据患者的临床表现、实验室检查结果和内分泌功能试验,糖尿病诊断成立,分型为特殊类型糖尿病中胰腺外分泌疾病,倾向遗传性胰腺炎,合并有周围神经病变和可疑糖尿病肾病1期的并发症,所以患者的诊断为:①胰源性糖尿病(遗传性胰腺炎)并周围神经病变和糖尿病肾病1期?②双眼白内障,③维生素D缺乏症,④混合痔。患者家系分析结果见表4。

患者家系分析结果
患者家系分析结果
| 检测基因 | 检测位置 | 检测方法 | 核苷酸变化 | 受检人 | 检测结果 |
|---|---|---|---|---|---|
| SPINK1 | ch5-147207583 | Sanger测序 | c.194+2T>C | Ia | 无变异 |
| Ib | 纯合变异 | ||||
| IIa | 杂合变异 | ||||
| IIb | 杂合变异 | ||||
| IIc | 杂合变异 | ||||
| IId | 杂合变异 | ||||
| IIe | 杂合变异 | ||||
| PRSS1 | Chr7-142460750 | Sanger测序 | c.623G>C | Ia | 无变异 |
| Ib | 杂合变异 | ||||
| IIa | 杂合变异 | ||||
| IIb | 无变异 | ||||
| IIc | 无变异 | ||||
| IId | 杂合变异 | ||||
| IIe | 无变异 |
注:先证者的爸爸(Ia)、先证者的妈妈(Ib)、先证者(IIa)、先证者的第一个妹妹(IIb)、先证者的第二个妹妹(IIc)、先证者的弟弟(IId)、先证者的第三个妹妹(IIe)
给予生活方式干预,限制脂肪、蛋白质摄入,补充微量元素与维生素,外源性胰酶制剂及胰岛素治疗,入院时五点血糖19.8-18.0-31.8-23.4-17.7 mmol/L,予门冬胰岛素4U 3U 3U三餐前皮下注射,甘精胰岛素6U睡前皮下注射,患者血糖一直比较高,逐渐增加胰岛素剂量,一周后五点血糖为17.4-11.2-23.6-20.4-13.5mmol/L,胰岛素调整为门冬胰岛素20U 14U 9U三餐前皮下注射,甘精胰岛素20U睡前皮下注射,患者血糖逐渐好转,偶有低血糖发生,再治疗一周后五点血糖为7.6-5.9-12.3-9.6-8.8 mmol/L,胰岛素调整为门冬胰岛素2U 10U 9U三餐前皮下注射,甘精胰岛素12U睡前皮下注射,患者病情好转出院
3个月后电话复诊患者仍予胰岛素治疗,偶有低血糖发生,胰岛素剂量调整为门冬胰岛素2U 6U 6U三餐前皮下注射,甘精胰岛素8U睡前皮下注射,空腹血糖7.0mmol/L左右。
胰源性糖尿病是指由于胰腺外分泌功能疾病导致的糖尿病,包括胰腺炎(急性、复发或任何原因引起的慢性胰腺炎)、胰腺切除术/创伤、肿瘤、囊性纤维化、血色素沉着症、纤维钙化性胰腺病。其中遗传性胰腺炎(HP)是一种少见的常染色体显性遗传疾病[2]。HP是一种常染色体显性遗传疾病,多于儿童期起病,发病年龄小,主要表现为反复发作急性胰腺炎,两代及以上家族中出现2例及以上胰腺炎患者,与过量饮酒,胆结石或创伤无关,伴随基因突变。HP疾病进程中可表现为反复腹痛、腹泻和胰腺内、外分泌功能不全的急性、复发性和慢性胰腺炎的相关临床表现,10%~25%的患者发展为糖尿病,糖尿病程度较轻[3]。HP的发病机制仍不明确,普遍认为与影响胰腺消化酶类活性的基因突变等遗传因素有关。目前缺乏HP确切的流行病学资料。HP的诊断依据为①自幼年即有反复的胰腺炎发作,②男女患病几率均等,③患者家族中至少还有另2例胰腺炎患者,④常出现胰管结石,⑤可以排除如酗酒、胆石、创伤、药物、感染及代谢紊乱等常见的胰腺炎诱因[4],尚需要和2型糖尿病,1型糖尿病MODY、Ig4相关性自身免疫性糖尿病、胰腺纤维钙化性糖尿病(FCPD)等相鉴别。其中胰腺纤维钙化性糖尿病是一种常发生于青少年的慢性非酒精钙化性胰腺炎。多见于热带发展中国家,其临床特征为发病年龄小,病程进展快,胰管内巨大结石。晚期表现为胰腺纤维钙化性糖尿病和或脂肪泻,易癌变。相关的报道多来自于印度、孟加拉国。其诊断标准是:①多发生于热带国家或地区,②符合WHO糖尿病诊断标准,③存在慢性胰腺炎的证据:腹部x线可见胰腺钙化或存在以下4点中的任意3点:i.自幼腹痛,ii.胰腺形态异常(超声、CT或内镜逆行胰胆管造影提示胰管扩张),iii.脂肪泻,iv.粪便糜蛋白试验提示胰腺功能异常,④除外酒精性或其他原因导致的慢性胰腺炎[5]。两者有很多相似之处,但从患者的遗传特点、地域等不支持该病的诊断。我们从这个家系还看到患者之间不同的个体临床表现也略有差异,说明基于基因突变的三维结构及重组胰蛋白酶的特性存在差异,这也提示我们需要进一步研究突变所致HP的确切机制。
1952年Comfort及Steinberg首次报道遗传因素与慢性胰腺炎有关[1],1996年遗传性慢性胰腺炎的致病基因位于7号染色体长臂,其阳离子胰蛋白酶原基因(PRSS1),阳离子胰蛋白酶原基因、阴离子胰蛋白酶基因(PRSS2),胰蛋白酶抑制剂基因(SPINKl),糜蛋白酶C基因(CTRC),囊性纤维化跨膜转导因子基因(CFTR),钙敏感受体基因(CASR)[6],2000年Witt等第一次发现丝氨酸蛋白酶抑制剂(serine protease inhibitor, Kazal type 1,SPINK1)的突变与慢性胰腺炎的发病有关,SPINK1位于7号染色体长臂(7q35),含5个外显子。SPINK1是在胰腺腺泡中合成的56个氨基酸的多肽,它与糜蛋白酶原同时合并成,糜蛋白酶原共同包裹在酶原颗粒中[7]。它是一种有效的蛋白酶抑制剂,可以在胰管内灭活胰蛋白酶的活性,起到保护腺泡的作用,是胰腺腺泡的第一道内在防御线。基因的内含子、外显子和启动子变异,可能引起其功能丧失或增强,也有可能不产生任何影响。c.194+2T>C突变常见于亚洲人群,突变导致剪切异常,SPINKl功能丧失性的突变导致抑制蛋白水平下降,所以胰蛋白酶激活不能被抑制,从而引起对胰腺炎的易感性增加[8]。
SPINKl和PRSS1基因的突变都分别是引起遗传性胰腺炎的病因,该家族患者存在两种基因的突变,临床上有很严重的胰腺钙化,但是胰腺炎症状却较轻,因此不同的突变体所致的临床症状可能有轻重之分,在基因突变的情况下,通常需要与其他基因的遗传突变和/或环境因素共同作用才有表现型。
HP患者患胰腺癌的风险增加50~70倍[9],可以肯定慢性胰腺炎是发生胰腺癌的高危因素,HP患胰腺癌的风险比其他类型的胰腺炎要高得多[10]。传统的胰腺导管腺癌的诊断方法是有限的,在10%的慢性胰腺炎患者、吸烟者和存在黄疸的情况下,肿瘤标志物被错误地升高,由于早期肿瘤的检测灵敏度低于50%,因此它的临床指导意义较小。CA19-9对早期胰腺癌(< 20mm,局限于胰腺)的检测灵敏度在纯胰液中仅为43%,其他50种肿瘤标志物,如CEA、SPAN-1和CA50,单独使用或联合使用,敏感性低于CA19-9,CT对小于2 cm的肿瘤和小于3 cm的肿瘤检测胰腺导管腺癌的敏感性分别为33%和50%[11]。
本例患者提示对于年龄小于25岁、体形消瘦、有腹痛、腹泻或者脂肪泻等临床表现,有糖尿病家族史,胰岛功能差但无酮症倾向、要高度警惕胰源性糖尿病,胰腺CT能显示胰腺的形态以及钙化程度,可作为常规的筛查手段,血糖波动较大的患者应加强膳食管理,尤其是限制脂肪、蛋白质摄入,补充微量元素与维生素,适当配合胃肠外营养。尽管与HP相关的致病基因不断被发现,但具体发病机制还未完全阐明,且不同的致病基因所存在的突变位点与HP的确切关系也未明确,对HP基因诊断及个性化的基因治疗还需要进行长期的研究。
所有作者均声明本研究不存在利益冲突

























