
患儿,女性,13天,试管婴儿。产检脐带绕颈2周,胎盘2级,经剖宫产娩出。生后1周无明显诱因出现嗜睡,气促,就诊于当地医院诊断为先天性心脏病,后症状逐渐加重伴皮肤瘀斑,为求进一步诊治转诊本院。入院完善相关检查诊断为:永存第五主动脉弓、主动脉缩窄、房间隔缺损、动脉导管未闭、二、三尖瓣关闭不全、新生儿肺炎。
嗜睡,哭闹减少,食欲减低,呼吸急促;皮肤瘀斑;口唇及甲床轻度发绀,双肺呼吸音稍粗,伴少量湿啰音,心前区无隆起,心率160次/分,律齐,心音可,未及明显杂音。肝肋下3指,质韧,无杵状指(趾),双下肢无水肿,囟门平软。
超声心动图、CT血管造影。
手术治疗(主动脉缩窄矫治术+房间隔缺损修补术+动脉导管切断缝扎术)。
患儿术后生命体征平稳,症状缓解,伤口甲类愈合,术后25天顺利出院。
超声医学科;心脏大血管外科;儿科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
永存第五主动脉弓(Persistent Fifth Aortic Arch,PFAA)是一种由于胚胎发育异常导致的罕见的主动脉弓部血管变异,主动脉弓同侧的第5弓动脉持续存在,于主动脉弓部构成双腔主动脉弓的特殊血管通道。1969年Van Praagh[1]首次报道并定义了PFAA,并描述了其特征。1973年Lzukawa报道首例存活的PFAA病例[2],由于该疾病罕见,均为散在病例报道,其发病率目前尚无流行病学数据。我们报告一例合并房间隔缺损、动脉导管未闭的重症PFAA新生儿,经过及时准确的诊断和有效的手术治疗后,该患儿在一个月内即顺利康复出院。本文旨在探讨此例PFAA诊治经验,从而提高对PFAA的认识,减少漏诊、误诊。
主诉:嗜睡伴气促5天。现病史:患儿为试管婴儿,产检脐带绕颈2周,胎盘2级。经剖宫产娩出,生后1周无明显诱因出现嗜睡,哭闹减少,食欲减低,伴呼吸急促,就诊于当地医院。查胸部CT示:双肺少许感染,具体诊治不详。后症状未见改善,伴皮肤花斑出现,为求进一步诊治,继而来本院门诊,门诊以"先天性心脏病"收入心脏大血管外科。既往史:平时健康状况:较差。个人史:经常居留地:湖北省宜昌市,放射性物质接触史:否认。家族史:父:健在,母:健在,子宫腺肌症病史。家族无遗传性及传染性疾病史。体格检查:患儿口唇及甲床轻度发绀,T:36.0℃,P:161次/分,R:54次/分,BP:82/47mmHg。神志嗜睡。心前区无隆起,心音正常,心率161次/分,心律整齐,未闻及杂音,胸肺听诊呼吸音异常,双肺呼吸音稍粗,伴少量湿啰音。无胸膜摩擦感。腹部外形膨隆,腹部触诊全腹柔软,肝肋下3指,质韧,无杵状指(趾),双下肢无水肿,囟门平软。
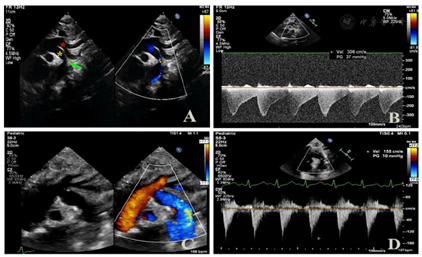
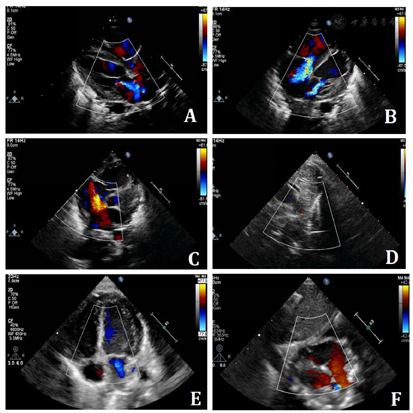
心电图提示:窦性心动过速;T波改变。床旁超声心动图:1.升主动脉于主动脉弓起始段(弓部内径约0.6cm)发出上位动脉弓和下位动脉弓两大分支,上位动脉弓分出头臂干、左颈总动脉、左锁骨下动脉后,多切面探查为盲端;下位动脉弓下行延续为降主动脉并降段狭窄,狭窄处内径约0.1cm,降主动脉起始段狭窄后扩张,内径约0.9cm。CDFI示降主动脉狭窄处血流速度增快,峰速3.1m/s,压差36mmHg(图1A、图1B);2.全心增大,左、右室室壁运动弥漫性减弱。另房间隔中部见一宽约0.66cm连续中断。二、三尖瓣形态、开放可,闭合不佳。CDFI:房间隔连续中断处见左向右为主的双向分流信号;降主动脉与左肺动脉见可见一细束左向右分流信号;二尖瓣口收缩期左房侧见中量反流信号;三尖瓣口收缩期右房侧见中至大量反流信号(图2A、图2B)。超声心动图显像提示:复杂先天性心脏病:1.主动脉弓发育异常永存第五主动脉弓(B型)并降主动脉缩窄;2.第四主动脉弓离断;3.房间隔缺损(左向右为主的双向分流);4.动脉导管未闭(即将闭合);5.二尖瓣中度关闭不全三尖瓣中至重度关闭不全;6.全心增大左、右室收缩功能测值减低;7.重度肺高压。
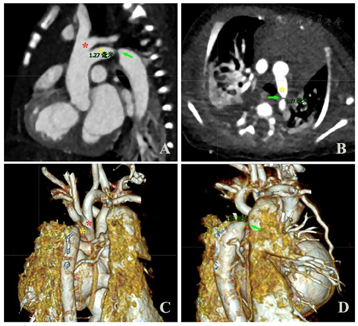
先心病CTA成像:检查所见:1.升主动脉管径约9.6mm。主动脉弓左弓左降。上弓发出头臂干、左颈总、锁骨下动脉;下弓弓降部局部管腔狭窄,管径约0.9mm,长1.5mm,其前后段管径分别约5.4mm、8.5mm,稍远较正常段宽7.9mm,降主动脉(穿膈段)宽6.7mm(图3)。2.房间隔中央部连续性中断,缺损区域大小8.6mm×9.4mm。3.右心房、室增大,右室心肌增厚。4.未见明确降主动脉发出较大侧枝血管影。提示:1.永存第五动脉弓(Weinberg B型),其中下支弓降部缩窄,狭窄后稍显扩张。2.房间隔缺损。3.右心增大;右室肥厚,肺动脉干增宽,超过同层面升主动脉,考虑肺动脉高压。
诊断:超声心动图是PFAA的首选影像学检查手段,经胸骨旁至胸骨上窝追踪扫查升主动脉及其弓部主要分支,通过对其解剖的多切面扫查可对PFAA进行初步诊断,但超声心动图扫查易受患者声窗的影响,且同时单平面显像难以显示弓部变异血管的全貌;CT血管造影及核磁共振可以多平面显示主动脉弓部血管解剖,三维重建后处理更能形象地显示主动脉弓部血管走形及其变异;经导管主动脉弓部造影是确诊的金标准,但因其为有创检查,临床应用受限,尤其是在年龄较小的患儿。
鉴别诊断:
C型PFAA与动脉导管未闭鉴别:动脉导管解剖位置在肺动脉与主动脉弓降部之间,而C型PFAA动脉导管缺如,PFAA起源于升主动脉。
与主动脉离断相鉴别:主动脉离断患者降主动脉通过动脉导管与肺动脉相连,主动脉弓部离断,不存在双弓结构。
患儿入院时病情危重,心源性休克并新生儿肺炎,入院后行强心、利尿、改善微循环、气管插管、辅助呼吸、化痰、抗感染等对症治疗。完善术前相关检查后行主动脉缩窄矫治术+房间隔缺损缝闭术+动脉导管切断缝闭术,延迟关胸并留置腹膜透析管,三天后行床旁关胸术,清理心包纵隔腔,未见活动性出血,温生理盐水冲洗后逐层关胸。继续呼吸机辅助、腹膜透析、抗感染、强心扩管利尿化痰,密观生命体征及内环境变化,并根据每日变化调整治疗方案。
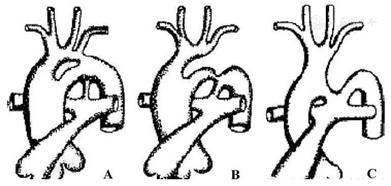
哺乳动物大血管发育过程中先后出现6对弓动脉,绝大多数情况下,双侧第5弓动脉大多在胚胎生命的第4周不形成或退化,当第5动脉共持续存在,即形成大血管水平的特殊通道,即PFAA[3]。Van Praagh、Gerlis等先后对PFAA进行报道[1,4]。Weinberg将该病分为三型:A型最常见,即双腔主动脉弓,弓上血管起源于上方的第四主动脉弓,伴或不伴发峡部狭窄;B型即永存第五对主动脉弓,第四主动脉弓离断或闭锁,第五弓动脉与降主动脉连接伴易行处狭窄;C型为体肺动脉连接型,无动脉导管[5,6](图4)。
PFAA常合并其他心血管畸形,包括动脉导管未闭、主动脉弓中断、肺动脉闭锁、法洛四联症以及大动脉转位等[7]。在一些遗传综合征病人中有该病表现,包括DiGeorge综合征,Corneliade Lange综合征以及PHACE综合征。单纯PFAA,血流动力学改变不明显,常无症状[8]。如合并主动脉缩窄,可增加左心室后负荷,下半身供血减少,高血压,上、下肢存在脉压差,最终导致充血性心力衰竭。如合并有动脉导管未闭,可导致肺循环血量增加,左心室前负荷增加,左室肥厚,进而肺动脉高压,右心功能衰竭。
超声心动图是诊断该病的首选检查方法,但受声窗和医师经验限制,仍有一定的误诊和漏诊率。在临床工作中,尤其合并心内结构畸形时,需辨别血流动力学与症状体征的不符合,应仔细扫查,特别是胸骨上窝的探查,以寻找病因。其次,由于本病罕见,对疾病的认识不足,可能仅诊断主动脉缩窄,而忽略双弓的存在。因此,仔细扫查胸骨上窝切面及加强理论知识的学习是必要的。血管造影术被认为是诊断PFAA金标准方法,但CT血管造影和心脏磁共振成像可以更精确地界定主动脉弓部解剖及其变异[9]。
PFAA与同侧的第四弓并存,两血管多处于同一切面,不会构成血管环而影响气道及食管,故如果PFAA无狭窄,一般不需处理。若合并主动脉弓缩窄,且对心功能有明显的影响,则需手术纠治[10]。手术方法有主动脉成形、第五弓动脉与降主动脉端端吻合、狭窄段人工血管置换以及经皮支架植入等[11]。本例合并先天性心脏病的B型PFAA重症患儿,术前心脏扩大,心功能损害并瓣膜反流,经过准确的术前影像学诊断、及时有效的治疗,患儿症状迅速缓解并在一月后顺利康复出院。
所有作者均声明本研究不存在利益冲突

























