
患儿(先证者)女,4个月23天,以"体重增长缓慢4个月"为代主诉入院。
全身皮下脂肪组织消失,毛发旺盛,有特殊面容:前额突出,耳廓稍大,三角脸,小下颌,舌体大、突出,腹膨隆,脐稍凸出,肝脏肋下平脐,质韧,脾脏肋下2 cm,四肢肌肉发达。
结合患儿病史、体格检查、实验室检查、二代测序结果提示BSCL2基因c.323T>G(p.Val108Gly)纯合变异,诊断明确。
低脂饮食,中链脂肪酸奶粉喂养,口服苯扎贝特降血脂,补充脂溶性维生素D和维生素E。
2周后复查肝功能谷丙转氨酶、谷草转氨酶、甘油三酯均较前上升,血常规、肾功能、心肌酶、总胆固醇正常,3个月后电话随访家长自诉肝脏较前进一步增大,质地较前变硬,4个月后复查腹部彩超提示肝大,剑突下达脐下约1 cm,右肋下达脐下约2 cm,肝实质回声光点细小,胆囊体积增大,大小约74 mm×18 mm,生化因脂血未予化验,患儿现11个月5天,体重7.5 kg、身长75 cm,会独站,会喊爸爸妈妈、说简单话语。
儿科;内分泌遗传代谢科;营养科;


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
先天性全身性脂肪营养不良(congenial generalized lipodystrophy,CGL),又称Berardinelli-Seip综合征,是一种罕见的常染色体隐性遗传病,该病多见于国外报道,发病率约为1/1 000万[1],目前尚无相关文献报道国内该病发病率,临床可表现为出生时或生后不久出现全身皮下脂肪组织消失、皮肤黑棘皮征、伴严重胰岛素抵抗甚至糖尿病、血甘油三酯升高、肝肿大、智力低下等特征。分别由AGPAT2、BSCL2、CAV1、PTRF基因引起CGL四种类型,其中CGL2是临床最常见且表型最严重的类型[2],早期诊断和治疗可改善患者预后。现回顾分析1例BSCL2基因变异导致CGL2,旨在提高对该病的认识,提高患者生活质量。
患儿女,4个月23天,以"体重增长缓慢4个月"为主诉于2021年12月21日入院。患儿出生体重3.0 kg,身长50 cm,生后半个月龄起发现体重增长缓慢,半月龄体重3.3 kg,身长不详,42天体重3.3 kg,身长54 cm,3月龄体重4.5 kg,身长57 cm,现4个月23天,体重5.8 kg,身长62.3 cm,伴全身脂肪组织逐渐消失、皮肤弹性差、全身多毛、肌肉发达(患儿自出生后临床特征见图1)。喂养方式:生后混合喂养,每日喂养总量不详(母乳量不详,奶粉量200~300 ml/d),无呕吐、腹泻等症状。42天体检时因体重增长缓慢添加低体重儿配方奶粉,效果差,体重每月增长不足1 kg。G2P2,孕39周剖宫产,3个月会抬头,现不会翻身,追视追听、逗笑可。父母非近亲结婚,均体健,有1哥哥,6岁,体健,家族中无类似疾病史。
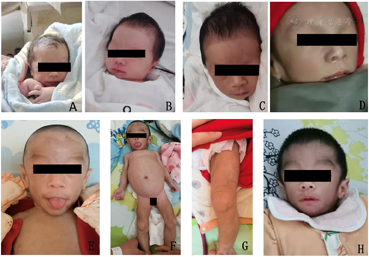
入院查体:体温36.6℃,心率122次/分,呼吸32次/分,身长62.3 cm(10 th),体重5.8 kg(<3 rd),体质量(BMI)14.94 kg/m2,头围39.9 cm(10 th),神志清,精神反应一般,全身皮下脂肪组织消失,皮肤弹性差,毛发旺盛。有特殊面容:前额突出,耳廓稍大,三角脸,小下颌,舌体大、突出。前囟平软,直径约1 cm×1 cm,口唇红润,咽无充血。颈软无抵抗,双肺呼吸音粗,心音有力,腹膨隆,脐部稍突出,肝脏肋下平脐,质韧,脾脏肋下2 cm,肠鸣音正常,四肢肌力正常,肌张力增高,四肢肌肉发达。外生殖器呈幼女型外观,阴毛PH1期。
A:出生;B:生后3天;C:生后20天;D:生后50天;E:4个月23天面部特征为前额突出、耳廓稍大、三角脸、小下颌、舌体大、舌突出;F:全身特征:全身皮下脂肪组织消失,腹部膨隆、腿部多毛、肌肉肥大;G:腿部多毛、肌肉发达;H:患儿8个月面部特征
血尿粪常规、血气分析、甲状腺功能、肝肾功能、心肌酶谱、电解质正常,血氨33.8 μmol/L(0~33 μmol/L),葡萄糖4.60 mmol/L(3.8~6.1 mmol/L),促肾上腺皮质激素10.16 pg/ml(7.2~63.3 pg/ml);皮质醇15.76 μg/dl(7.26~32.28 μg/dl),胰岛素9.220 μIU/ml(2.6~24.9 μIU/ml),C肽4.34 ng/ml(1.1~4.4 ng/ml);胰岛素样生长因子39.06 ng/ml(56~344 ng/ml);血脂:总胆固醇4.36 mmol/L(3.6~5.69 mmol/L),甘油三酯15.92 mmol/L(0.38~1.7 mmol/L),高密度脂蛋白0.35 mmol/L(0.8~1.7 mmol/L),低密度脂蛋白1.69 mmol/L(1.9~3.6 mmol/L),载脂蛋白A1 0.64 g/L(1~1.6 g/L),载脂蛋白B 1.02 g/L(0.6~1.1 g/L),心脏彩超:卵圆孔未闭,室间隔及室壁稍厚,心包积液(左室侧壁处厚约3.3 mm,右室侧壁处厚约3.6 mm)。心电图检查示⑴窦性心律;⑵ST-T改变。腹部彩超(肝胆脾胰)提示肝大、脾大,肝实质回声稍增密增强,双肾及肾上腺彩超未见明显异常,血氨基酸酰基肉碱谱、尿有机酸分析未见明显异常。
为明确病因,获得患儿父母知情同意后并签署书面同意书,并经伦理委员会审查批准后(伦理号2022-K-026),采集患儿及父母外周血标本各2ml,EDTA抗凝,从受检者样本中提取基因组DNA,构建基因组文库。通过探针杂交捕获目标基因(约20 000个)外显子及毗邻剪接区域(约20bp),并进行富集。对富集的基因进行质量控制,利用高通量测序仪进行测序。测序原始数据首先去除不符合质控要求的reads,然后运用BWA软件与UCSC提供的hg19版本人类基因组参考序列进行比对,经过GATK的HaplotypeCaller找出其中的SNV和InDel变异,并对目标区域的覆盖度和测序质量进行评估。依据严格的筛选标准对变异进行过滤,并添加HGMD数据库、蛋白功能预测软件的相关注释信息。对明确或可能与受检者临床表型相关的基因变异,采用Sanger测序进行验证。对于疑似变异,参照美国医学遗传学和基因组学会(American College of Medical Genetics and Genomics, ACMG)评级指南对其致病性进行评估。
全外显子测序显示,患儿BSCL2基因存在c.323T>G(p.Val108Gly)纯合变异,父母均携带(杂合),该变异为HGMD与gnomAD数据库未报道过的变异,根据ACMG指南评估为可能致病性变异。
CGL2型诊断明确。
获得性全身型脂肪营养不良:也称为Lawrence综合征,多在儿童期出现,最初为局部皮下脂肪丧失后发展为全身性脂肪丧失,少数呈现腹壁和骨髓脂肪丢失,发病机制不明确,1/4患者起病时为脂膜炎表现,另外1/4患者可伴有自身免疫疾病,如类风湿关节炎、系统性红斑狼疮等,另1/2患者为特发性,不伴脂膜炎及自身免疫性疾病。部分患儿会有胰岛素抵抗、高甘油三酯血症、脂肪肝,甚至糖尿病。支持点:该病也可表现为全身性脂肪丧失,可合并胰岛素抵抗、高甘油三酯血症、糖尿病等,不支持点:起病年龄不同,CGL不伴脂膜炎及自身免疫性疾病,基因检测结果不支持。
家族性部分性脂肪营养不良:该病根据致病基因不同,可分为5种亚型,患者脂肪丢失几乎可发生在生长发育任何阶段,从幼年至成年均可发生。遗传方式可以是常染色体显性遗传、常染色体隐性遗传或伴性遗传。临床表现为四肢、腹部和胸部对称性脂肪缺失,肌肉组织突显,而下颌、锁骨上、腹腔脂肪堆积,出现类库欣综合征面容。支持点:可出现脂肪消失,不支持点为脂肪消失为对称性脂肪缺失,局部脂肪堆积,可出现类库欣综合征面容,基因检测结果不支持。
低脂饮食,中链脂肪酸奶粉喂养,针对高甘油三酯血症,贝特类药物为治疗首选药物。由于此类药物尚无应用于儿童的疗效及安全性研究,且根据《儿童青少年血脂异常防治专家共识》没有被推荐为儿童及青少年常规降脂药物[3],《甘油三酯增高的血脂异常防治中国专家共识》提出甘油三酯增高,应积极改善生活方式,若甘油三酯≥5.65mmol/L,应立即启动药物治疗[4]。与家长沟通后同意应用并签署知情同意书,给予口服苯扎贝特50mg每天一次降血脂治疗,严密观察不良反应,同时口服脂溶性维生素D 400U/d、维生素E 5mg每天一次等对症支持治疗。
2周后当地医院复查肝功能谷丙转氨酶116 U/L(7~30 U/L),谷草转氨酶75 U/L(14~44 U/L),甘油三酯15.92 mmol/L(0.45~1.7 mmol/L),均较前上升,血常规、肾功能、心肌酶、总胆固醇正常,3个月后电话随访家长自诉肝脏较前进一步增大,质地较前变硬,4个月后复查腹部彩超(肝胆脾胰肾)提示肝大,剑突下达脐下约1 cm,右肋下达脐下约2 cm,肝实质回声光点细小,胆囊体积增大,大小约74 mm×18 mm,生化因脂血未予化验,患儿现11个月5天,体重7.5 kg、身长75 cm,会独站,会喊爸爸妈妈、说简单话语。
CGL2是由BSCL2基因变异引起,该基因位于11q13染色体上,长度17.12kb,包含12个外显子,编码含389个氨基酸的seipin的跨膜蛋白,属于内质网固有蛋白,有助于将蛋白质、脂肪和其他分子运输到细胞内外的特定位点,该蛋白在脂肪组织、神经系统和睾丸中高表达,通过限制在非脂肪细胞中脂肪形成和脂滴堆积,在脂肪过剩时促进脂肪细胞生成,从而达到脂质的动态平衡。若BSCL2基因变异时脂肪细胞生成及成熟障碍,使脂质扩散到细胞及其周围组织,进而引发一系列损伤[5]。
截至2021年4月,在HGMD中检索BSCL2基因,共报道49种突变,包括22种错义/无义突变,9种剪切突变,微缺失8种,微插入5种,微插入缺失2种,大片段缺失2种,复杂重排1种,变异频率较高的位点为c.782dupG、c.545-546insCCG、c.669insA、c.280C>T、c.974dupG、c.659delGTATC、IVS4+1G>A与c.565G>T均为移码变异或无义变异,导致患儿发生严重并发症的多为无义变异[6]。本例患儿为进一步明确病因,完善全外显子检测提示BSCL2基因c.323T>G(p.Val108Gly)纯合变异,父母均携带(杂合),符合常染色体隐性遗传,该变异为未报道的变异,按照ACMG变异分类标准,为可能致病性变异,结合患儿病史、临床表型及辅助检查,符合CGL2型,故CGL2型诊断明确。
CGL2主要的临床特点为出生或出生后不久出现的病理性的几乎全部的脂肪组织缺失,出生后、婴儿期和儿童早期可出现脂质代谢异常和糖代谢异常,脂质可异常分布于肝脏、血液、心脏,引起肝大、脂肪肝、肝硬化、高甘油三酯血症、肥厚性心肌病等,若进一步加重,可导致反复急性胰腺炎、高胰岛素血症、胰岛素抵抗相关糖尿病,黑棘皮作为胰岛素抵抗表现,常分布在颈部、腋窝、腹股沟等部位,在儿童早期就可出现。CGL2患儿的食欲旺盛,生长迅速,部分患儿可伴有脐疝、脾肿大,青春期也可表现为肢端肥大,女性患儿中普遍存在轻度多毛症、阴蒂肥大和月经不调,部分患者甚至患有原发性或继发性闭经和多囊卵巢[6,7]。CGL2的男性可表现为无精症和性腺功能减退,主要是由于病变蛋白在性腺组织中的重要功能作用[8]。除了以上临床特点,由于Seipin在神经系统中高表达,部分患儿可伴有不同程度的智力发育落后,但具体分子机制尚不明确[9]。国内赵玲霞等[10]对既往23篇文献报道96例BSL2基因突变导致的CGL2患儿临床表现进行总结,皮下脂肪菲薄100%,三酰甘油血症90.6%,血清谷丙转氨酶升高81.5%,单纯肝大73.8%,高胰岛素血症71.1%,黑棘皮症66.7%,空腹血糖升高55.6%,智力障碍55.4%,心肌病28.1%,肝大、脾大16.4%。本研究报道患者出生时正常,生后逐渐出现全身皮下脂肪萎缩、特殊面容、腹部膨隆、多毛等表现,实验室检查高三酰甘油血症、肝脾肿大表现,腹部彩超提示脂肪肝,结合二代测序结果,诊断CGL2明确。Haghighi等[2]总结了CGL2的临床症状,主要临床症状包括躯干、四肢及面部脂肪萎缩;肢端肥大症(巨人症、肌肉肥大、骨龄超前、手脚增大、阴蒂肥大或男性外生殖器肥大);肝大;血清三酰甘油升高和(或)伴有高胆固醇血症;胰岛素抵抗(血清C肽与胰岛素水平升高);黑棘皮症,次要临床表现包括肥厚型心肌病;精神运动发育迟缓或轻至中度智力障碍(智商为35~70);多毛症;女性性早熟;8%~20%患儿存在骨囊肿;静脉曲张,便于临床早期识别。
目前尚无治愈CGL2的特效方法,但早期诊断可预防严重并发症或死亡,对症治疗改善患儿的生存质量。应根据患儿临床表现及疾病严重程度采取不同治疗策略。饮食管理和运动治疗是治疗CGL的基本方法,需保证足够的能量以维持其正常生长和发育,鼓励患儿低脂饮食,限制饱和脂肪酸摄入,若合并糖尿病,还应适当限制碳水化合物的摄入量。药物治疗方面,应用时应慎重,注意药物的疗效、年龄限制、适应证及安全性研究。二甲双胍可改善胰岛素抵抗,起始剂量为10mg/(kg·次)每天2次,但若患者合并糖尿病时,大剂量胰岛素为主要的降糖药物。苯扎贝特是贝特类调制药物,可参与脂代谢过程并对转录因子活性进行调节,能通过线粒体脂肪酸β氧化途径进而对脂肪酸摄取、转化、分解过程产生影响[11],剂量为3~5mg/(kg·次)每天2次,治疗过程中应根据三酰甘油水平及体重调整剂量,部分患儿需联合他汀类药物有利于降低低密度脂蛋白胆固醇水平,大剂量的ω-3不饱和脂肪酸5~10g/d,补充维生素E有助于减少肝脂肪变性[12]。定期复查血尿常规、肝肾功能、心肌酶谱、血脂,同时密切监测相关并发症发生。对于无脂代谢和糖代谢患儿,建议定期随访即可。对于合并胰岛素抵抗、糖尿病或高甘油三酯患儿,若病情较轻,建议饮食控制及运动锻炼,每日摄入基础热量为25~35kcal/kg,大部分患儿在早期就出现多种代谢并发症,常需要饮食、运动、降糖药及降脂药等多方面综合治疗[12]。2014年美国食品药品监督管理局(FDA)批准,重组人瘦素类似物美曲普汀作为饮食和生活方式改变的辅助手段,可用于治疗成人和儿童脂肪营养不良的代谢并发症[13]。Brown等[14]应用美曲普汀的平均剂量为0.082 mg/(kg·d)[范围0.04~ 0.19 mg/(kg·d),最大剂量4.1 mg/d],治疗后糖化血红蛋白及三酰甘油水平均有明显下降,改善食欲控制、胰岛素敏感性和肝脏脂肪变性,治疗最常见的不良反应是低血糖、头痛、恶心、体质量减轻和腹痛等。
CGL2远期生活质量和预后均较差[15],因此,对于临床上具有自幼出现皮下脂肪萎缩、肝肿大、高甘油三酯血症、黑棘皮症等典型症状患者,应警惕CGL2的可能,尽早进行基因检测以明确诊断及分型,避免漏诊误诊。对于有阳性家族史,再孕应行遗传咨询。对于明确诊断患者,应针对病情给予个体化治疗,尽早给予低脂饮食,及早纠正代谢紊乱及防治并发症以改善患者预后,提高患者生活质量。
所有作者均声明本研究不存在利益冲突

























