
丙酮酸脱氢酶复合物缺乏症(pyruvate dehydrogenase complex deficiency disease,PHD)是一种线粒体能量代谢异常的遗传性疾病,基因突变引起丙酮酸脱氢酶复合物(pyruvate dehydrogenase complex,PDHc)活性下降,导致丙酮酸代谢障碍,造成机体能量生成不足和乳酸堆积,进而出现神经系统结构和功能损害。PHD婴幼儿期发病多见,临床表现复杂,可出现持续高乳酸血症、间歇性肌无力或共济失调、运动发育迟缓、慢性神经病变等[1]。国内仅有少量个案报道[2,3,4]。本文回顾性分析1例PHD患儿的临床表现、实验室检查和头颅MRI特征、家系基因分析及诊治过程,以期为该病的诊治提供依据,报道如下。
丙酮酸脱氢酶复合物缺乏症(pyruvate dehydrogenase complex deficiency disease,PHD)是一种线粒体能量代谢异常的遗传性疾病,基因突变引起丙酮酸脱氢酶复合物(pyruvate dehydrogenase complex,PDHc)活性下降,导致丙酮酸代谢障碍,造成机体能量生成不足和乳酸堆积,进而出现神经系统结构和功能损害。PHD婴幼儿期发病多见,临床表现复杂,可出现持续高乳酸血症、间歇性肌无力或共济失调、运动发育迟缓、慢性神经病变等[1]。国内仅有少量个案报道[2,3,4]。本文回顾性分析1例PHD患儿的临床表现、实验室检查和头颅MRI特征、家系基因分析及诊治过程,以期为该病的诊治提供依据,报道如下。
患者,男,12岁,以"四肢无力10 a余,加重11 d"为主诉于2017年9月12日入院。10 a前患儿因"感冒"后出现双下肢无力,不能独自行走,休息15 d后症状缓解,可正常活动,但不耐受疲劳。2a前,患儿剧烈活动后出现肢体无力,行走不稳,就诊于当地医院,基因检查提示"PHD可能",未行特殊治疗。11 d前患儿受凉后发热,体温波动于37~38 ℃,伴咳嗽,痰不易咳出,并出现双上肢上抬困难。3d前患儿四肢无力进一步加重,双上肢不能上抬,双腿不能站立,并出现呼吸困难症状,急诊住院治疗。患儿系第1胎足月自然分娩,生后无窒息缺氧病史,父母非近亲结婚,均体健,有1弟体健。
入院时体温36.0 ℃,脉搏63次/min,呼吸26次/min,血压96/59 mm Hg (1 mm Hg = 0.133 kPa),身高155 cm,体质量30 kg。急性病容,表情痛苦,咽部黏膜充血,扁桃体Ⅰ~Ⅱ度肿大,双肺呼吸音粗,可闻及痰鸣音。神经系统查体:神智清,精神差,言语欠清晰,高级智能大致正常,双侧上睑下垂,眼球活动无异常,对光反射正常,余颅神经正常,全身深浅感觉正常,颈屈肌力2级,双上肢肌力2级,双下肢肌力3级,四肢肌张力减弱,四肢腱反射减弱,髌阵挛、踝阵挛阴性,双下肢病理征阴性,双手指鼻试验、一字步及闭目站立征不能完成。脑膜刺激征阴性。
入院后即行实验室检查、电生理检查、影像学检查等,并对患儿及家族成员线粒体病相关核基因突变情况进行检测分析。结果显示:患儿血清C反应蛋白144.5 mg/L,血沉36 mm/h,降钙素原定量0.57 μg/L,静息血乳酸波动在3.6~5.6 mmol/L,均增高。心电图、脑电图未见特异性改变;肌电图提示四肢多发周围神经病变;四肢体感诱发电位显示深感觉传导通路轻度异常;胸部CT提示双肺炎症。患儿2a前于外院头颅MRI显示双侧苍白球对称性条状长T1长T2信号,FLAIR、DWI高信号;本次入院头颅MRI显示双侧大脑脚、豆状核对称性片状长T1长T2信号影,FLAIR、DWI高信号影(图1)。基因检测发现,患儿PDHA1基因存在半合子突变,c.376 C > T (p. Arg126Cys)。患儿母亲存在PDHA1基因该位点的杂合突变,其父亲、弟弟、姨、舅舅均未发现该基因突变。该变异符合X连锁遗传方式,其母为变异携带者。
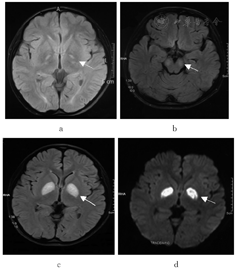
注:a为2a前头颅MRI影像,示双侧苍白球对称性FLAIR序列高信号;b-d为本次就诊时头颅MRI影像,b显示双侧大脑脚对称性FLAIR序列高信号,c-d显示双侧基底节区豆状核对称性FLAIR和DWI序列高信号。
患儿主要表现为发作性四肢无力,血乳酸增高,本次加重为呼吸道感染诱发,伴呼吸困难,头颅MRI示双侧脑干及基底节区对称性病变。PDHA1基因检测提示376 C>T突变,确诊为PHD。入院后给予抗感染、祛痰止咳及其他对症治疗,针对该病给予维生素B1 300 mg/d,左卡尼汀2 g/d,辅酶Q10 100 mg/d,艾迪苯醌45 mg/d,硫辛酸100 mg/d,给予生酮饮食,即高脂、限制糖类及淀粉类等高碳水化合物、适当蛋白质饮食。住院治疗1个月患儿病情稳定,无呼吸困难症状,上睑下垂较入院时有所减轻,但仍存在反应迟钝,少言语,双上肢肌力恢复至3级,双下肢肌力恢复至4级,可独自站立,行走需搀扶。出院后继续维持该治疗1个月,症状无加重,上睑下垂症状有好转,言语稍增多,反应迟钝,可独自进餐,行走仍需搀扶。
PHD最早于1970年被Blass等[5]报道,我国对该病的认识始于2007年张尧等[2]通过PDHA1基因分析确诊的1例男性患儿。PHD多引起神经系统和肌肉的损害,个体差异大,胎儿期至成人各年龄阶段均可发病,主要表现有先天性乳酸酸中毒、脑发育异常、婴儿痉挛症、间歇性肌无力或共济失调、慢性神经病变等。头颅MRI表现包括胼胝体发育不良,基底节和中脑对称性病变[1]。本例患儿2岁发病,表现为发作性肌无力,多由感染、发热诱发加重,与相关报道一致[3]推测可能为急性能量耗竭和酸中毒加重造成脑、肌肉损伤。实验室检查提示持续性高乳酸血症,肌电图提示多发周围神经病变,头颅MRI显示双侧大脑脚、豆状核对称性片状异常信号,基因检测发现患儿PDHA1基因发生半合子突变,其母为携带者。结合患儿临床表现、头颅MRI及基因检测结果可明确诊断为PHD。
PDHc是线粒体能量代谢途径中一组重要限速酶[6],PDHc缺乏时,丙酮酸代谢成乙酰辅酶A途径受阻,仅能经乳酸脱氢酶催化生成大量乳酸从而导致乳酸酸中毒,由于乙酰辅酶A缺乏,三羧酸循环途径受阻,三磷酸腺苷生成不足,而脑和肌肉均为能量高需求组织,脑损伤和肌无力或运动不耐受、肌张力低下常见。PDHc中丙酮酸脱氢酶α亚基缺陷是PHD最常见原因,约占70%,由PDHA1基因突变所致,PDHA1基因位于X染色体Xp22.1~22.2,该病为X连锁隐性遗传性疾病[1, 7]。国外有研究[8]显示多数PDHA1基因发生自发突变,仅少数突变来自母亲。本例患儿PDHA1基因检测发现与其母存在同一位点突变。PDHA1基因突变的女性携带者因X染色体的随即失活导致不同组织中突变基因和正常基因的数量不同,而表达正常丙酮酸脱氢酶α的比例影响体内残余PDHc的活性[9],女性患者多有癫痫发作,可不伴高乳酸血症,该患儿母亲尚无症状,但对其仍应定期进行随访。PHD目前仍缺乏特效治疗,预后差,随病情进展,多数患者在儿童期死亡。生酮饮食,即严格控制碳水化合物的摄入可减少丙酮酸的生成,进而缓解乳酸酸中毒;高脂肪饮食可通过脂肪酸产生乙酰辅酶A从而缓解三磷酸腺苷生成不足,改善临床症状[10]。PDHc还包括多种辅酶因子,如焦磷酸硫胺素、硫辛酸、辅酶A、Mg2+等。补充硫胺素和硫辛酸等可改善线粒体功能[1]。PHD患儿早期给予生酮饮食可降低血乳酸水平,大剂量维生素B1、左旋肉碱及硫辛酸可改善神经系统症状和线粒体功能,提高生存质量[3,4]。Ferriero等[11]研究发现,苯丁酸可通过抑制丙酮酸脱氢酶激酶的磷酸化作用从而增强PDHc活性,在动物模型中已取得预期结果,但对人体应用的有效性和安全性有待进一步研究证实。本例患儿经足量维生素B1、左旋肉碱、硫辛酸、辅酶Q10及艾迪苯醌的治疗,辅以生酮饮食,病情趋于稳定并有好转趋势。说明PHD临床表现复杂,头颅MRI及基因检测对疾病诊断有重要价值,早期合理治疗可稳定病情。但由于随访时间短,该治疗方案的疗效评估尚需进一步随访。

























