
患儿,女,5岁,以"间断发热、腹痛、腹泻2个多月"为代主诉收入院。
患儿2个月前无明显诱因出现间断发热、腹痛等不适,伴腹泻、里急后重,不伴咳嗽、呕吐、便血、皮疹等。查体:脐周压痛,肛周截石位3点钟方向可见一肛周脓肿。
临床症状主要表现为间断不明原因发热、反复口腔黏膜及生殖器黏膜溃疡、肛周脓肿、消化道溃疡、腹痛、腹泻等表现;电子结肠镜提示全结肠多发溃疡;基因检测示:TNFAIP3基因突变(c.866delA),导致p.H289Pfs*3,造成移码突变。结合临床症状、辅助检查及基因检测结果共同诊断A20单倍剂量不足。
给予肠内营养、英夫利西单抗静脉输注治疗。
患儿目前病情稳定,未再出现不明原因发热、腹痛、腹泻等症状,但口腔黏膜及生殖器黏膜溃疡易反复,肛周脓肿无明显变化。
儿科;消化内科;遗传代谢科;妇产科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
A20单倍剂量不足(haploinsufficiency of A20,HA20)是一种常染色体显性遗传病,由肿瘤坏死因子α诱导蛋白3(tumor necrosis factor α-induced protein 3,TNFAIP3)突变引起,属于单基因自身炎症性疾病的一种。其特征是多器官的炎症反应和自身免疫反应[1]。HA20临床特征异质性较大,以反复口腔、生殖器及胃肠道溃疡等表现较为常见[2,3]。不同的TNFAIP3基因突变位点与临床症状的异质性密切相关,因此明确TNFAIP3基因的突变位点,对于明确诊断、治疗方案的选择及评估预后至关重要。现回顾性分析1例以间断发热、腹痛、腹泻为主诉的患儿临床资料及遗传学特点等。
患儿,女,5岁,以"间断发热、腹痛、腹泻2个多月"为主诉于2021年10月15日入院。2个月前无明显诱因出现间断发热,热峰39.3℃,伴腹痛、腹泻,腹痛以脐周为主,大便7~8次/d,呈黄稀糊或稀水样便,含黏液,伴里急后重,无便血,在当地诊所给予三代头孢抗感染治疗半个多月,体温及腹泻均无明显缓解。追问病史,既往1岁左右开始反复出现无明显原因发热、腹泻、口腔及生殖器溃疡,未正规诊治。
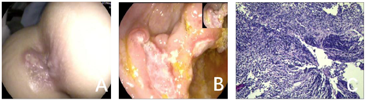
体格检查:发育正常,呼吸稍急促,全身皮肤黏膜无黄染、皮疹及出血点,全身浅表淋巴结未触及肿大,口唇稍干燥,咽无充血,颈软,无抵抗,心肺听诊无异常,肝、脾肋下未触及,腹软,未触及异常包块,脐周压痛,肛周截石位3点钟方向可见一红色肿物表面破溃(图1A)。神经系统检查无异常。
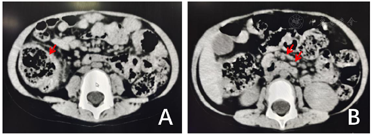
辅助检查:入院查血常规,白细胞计数14.84×109/L,血红蛋白86 g/L,血小板计数396×109/L,中性粒细胞百分比57.2%,淋巴细胞百分比31.1%;C反应蛋白166.68 mg/L,红细胞沉降率53 mm/h,降钙素原4.07 ng/ml,血尿便细菌培养、血生化、凝血功能、血尿便培养均阴性,脑脊液化验、EB病毒、巨细胞病毒检测、骨髓涂片及培养、风湿因子、自身抗体谱、结核菌素T细胞检测均为阴性,腹部彩超无明显异常。全腹部CT:1.右下腹网膜增厚,邻近肠管扩张;2.中下腹肠系膜淋巴结明显增多,部分肿大,部分沿肠系膜上动脉周围分布走行(图2)。
结合患儿病程长、外院应用三代头孢菌素抗感染治疗1个月无效、血常规及炎症指标仍高,考虑耐药菌感染可能性大,经院内特殊级抗菌药物审核专家组会诊后给予超广谱抗菌素美罗培南针抗感染治疗,患儿体温恢复正常,但腹痛、腹泻无明显好转。进一步完善电子结肠镜(图1B),提示全结肠可见多处不规则深溃疡,肛周可见脓肿。镜下诊断:1.结肠黏膜溃疡性改变(克罗恩病?免疫缺陷病?);2.肛周脓肿。黏膜病理(图1C):1.回肠末端、回盲部、升结肠、横结肠、降结肠、直肠黏膜慢性炎症;2.免疫组化结果显示,CD10(灶状+),CD20(B区+),CD3(T区+),CD43(+),CD5(灶状+),CD7(灶状+),CD79α(B区+),Ki-67(生发中心90%+),TdT(-),抗酸(-)。根据患儿病史、症状、体征及辅助检查,初步诊断:克罗恩病?免疫缺陷病?
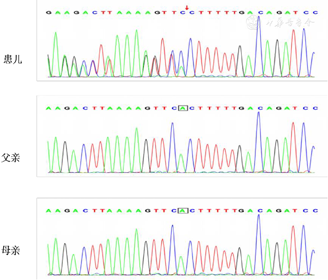
基因检测:同家长沟通后治疗上给予沙利度胺口服,3周后因出现肢体麻木、震颤等副作用停止使用。同时基因重排结果回示排除淋巴瘤。进一步沟通并签署知情同意书后,取患儿及其父母的新鲜全血2 ml,利用家系全外显子测序分析,发现患儿存在TNFAIP3基因杂合变异(NM_001270508:c.866delA),为移码突变。该变异位点经Sanger测序验证(图3),可见患儿TNFAIP3基因杂合变异(c.866delA),而其父母均为野生型,为自发突变。HGMD及Clinvar数据库均无该变异位点的相关报道和致病性分析。根据美国医学遗传学和基因组学学会指南,该变异判定为致病性变异,证据为致病性证据超强PVS1(LOF变异导致基因功能可能丧失)、致病性证据中等PM2(在正常人群数据库中的频率为低频变异)、致病性证据中等PM6(假定新发变异)。
患儿既往反复出现无明显原因发热、腹泻、口腔及生殖器溃疡等症状,结肠镜检测可见全结肠多处深大溃疡,综合患儿临床表现、结肠镜及基因检测结果,HA20诊断明确。
该病需与以下疾病相鉴别:(1)白塞病。白塞病是一种病因未明的慢性、反复发作性全身血管炎症性疾病。以复发性口腔溃疡、生殖器溃疡、眼炎和皮肤损害为常见的临床特征,也可累及多个系统的慢性全身性血管炎性疾病。该病实验室检查无特异性指标,通过基因检测可与HA20相鉴别。(2)肠结核。一般可有肠外结核病史,可出现低热、消瘦、腹痛、腹泻、便秘、便血、腹部包块等症状与体征,PPD实验及结核菌素实验有助于诊断,结肠镜黏膜病理可见干酪样肉芽肿,但检出率较低,基因检测可排除该疾病。
积极沟通后,监护人拒绝应用糖皮质激素,签署治疗同意书后给予"英夫利西单抗(类克)"按照指南静脉治疗。
目前已正规治疗近半年,未再出现无明显诱因发热、腹痛、腹泻等症状,但口腔黏膜及生殖器黏膜溃疡易反复,肛周脓肿如前,病情稳定,已正常入学。
A20是一种由TNFAIP3基因编码的蛋白质,它是NF-κB信号通路的负调节因子,在上皮细胞、树突细胞、T淋巴细胞和B淋巴细胞中均有表达,在机体免疫、细胞凋亡、炎症和癌症等多种生理和病理过程中发挥着重要作用[4,5,6]。A20蛋白由一个N端OTU结构域[氨基末端卵巢肿瘤区结构域(ovarian tumor domain,OTU)]和一个包含7个锌指结构域的C端结构域[羧基末端锌指区(zinc finger domain,ZnF)]组成[7,14]。OTU结构域可以通过调节IKK激酶复合物的调节亚基来拮抗NF-κB必需调节因子上游的泛素化,进而抑制NF-κB的激活,调控机体多种细胞活动[5]。而ZnF结构域是一种泛素连接酶,可引起受体相互作用蛋白中Lye8的泛素化和降解,从而辅助抑制NF-κB的活化[8]。
A20蛋白的缺失会导致NF-κB通路异常激活,引起或参与多种自身免疫性和自身炎症性疾病的发生[9]。迄今为止,已报道了43种A20的致病变异。以"haploinsufficiency of A20"和"TNFAIP3"为关键词检索PubMed数据库(截至2022年5月14日),共检索到53篇相关文章,涉及40个家系的72例患者,其中男28例,女44例,男女比例约为1∶2,平均发病年龄约为5.3岁。以"HA20"为关键词检索中国知网数据库,未检索到相关文献。以"HA20"为关键词检索中国知网数据库,仅检索到4篇病例报道[15,16,17,18]。已报道的HA20患者的临床症状多样,多数患者有反复口腔黏膜和生殖器黏膜溃疡、消化道溃疡、红斑结节、毛囊炎样皮疹、血栓性静脉炎、皮疹、腹痛、腹泻等症状,部分患者出现关节炎、桥本甲状腺炎、自身免疫性肝炎、系统性红斑狼疮、间质性肺炎、无菌性脑膜炎、霍奇金淋巴瘤、IgA血管炎、慢性肝炎、肾病综合征等胃肠外表现,甚至出现IgG2和IgG4缺乏、全血细胞减少和反复感染等[10,11]。
HA20的临床表现具有地域差异,东亚患者中以反复不明原因发热症状居多,而其他地区以反复口腔黏膜及生殖器黏膜溃疡症状较为常见[12];东亚地区的HA20患者与其他地区相比,不易合并如SLE和自身免疫性甲状腺疾病等自身免疫性疾病[12]。检索已发表报道显示,TNFAIP3基因突变位点不同可致临床症状具有较大的异质性,如p.His577Alafs*95突变患者多有关节炎及桥本甲状腺炎症状;c.1804A>T,p.T602S突变多有反复发烧和持续感染,伴有口腔溃疡、皮疹、眼结膜炎、肛周溃疡和肠道炎症等症状;c.1777C>T,p.Q593X和c.2126A>G(p.Gln709Arg)突变可有系统性红斑狼疮表现;c.1906C>T突变可致成人Still病。本报道中患儿TNFAIP3基因存在突变(c.866delA),从而引起p.H289Pfs*3,造成移码突变,该位点突变的致病等级较高。患儿的临床资料提示该位点的突变有发病年龄较早、症状较重的特点,临床症状主要表现为间断不明原因发热、反复口腔黏膜及生殖器黏膜溃疡、肛周脓肿、消化道溃疡、腹痛、腹泻等表现,英夫利西单抗治疗有效,预后暂不明,需继续随访。
目前HA20尚无统一诊断标准。其临床症状差异性较大,实验室检查指标无特异性,急性期可见血沉、C反应蛋白、部分炎症因子及自身抗体等升高,然而这些指标并不特异,因此极易造成漏诊、误诊,从而延误治疗。目前全外显子测序正越来越广泛地用于遗传性单基因自身炎症性疾病的诊断,临床患者发病年龄较小,出现反复发热、反复口腔溃疡、生殖器及胃肠道溃疡,甚至以上提及的所有临床症状,或出现不能解释的化验指标异常、胃肠外表现等症状的患者,都需进一步完善基因检测以明确诊断。
HA20的治疗目前尚无国际统一标准方案。主要的治疗药物有糖皮质激素、免疫抑制剂和生物制剂。治疗方案的选择需结合患者的年龄、临床症状及基因突变位点,目前多认为大多数患者急性期对糖皮质激素是敏感的,然而长期大剂量糖皮质激素会引起股骨头坏死、严重电解质紊乱、影响儿童生长发育等一系列并发症,因此使用的剂量及时间需权衡斟酌。目前可应用的免疫抑制剂有秋水仙碱、沙利度胺、硫唑嘌呤、环孢素、甲氨蝶呤、霉酚酸酯、胱抑素C等,但大多需要联合糖皮质激素来控制症状[13]。生物制剂主要包括抗TNF-α(英夫利西单抗、阿达木单抗)、IL-1受体拮抗剂(阿那白滞素)、IL-6受体拮抗剂(托珠单抗)、JAK抑制剂(托法替尼)等,可有效抑制HA20患者的全身炎症反应[14]。再次回顾本报道中的患者,该患儿发病年龄较小,临床症状有无明显诱因反复发热、反复口腔溃疡、生殖器溃疡及胃肠道溃疡等表现,实验室检查可见急性炎症指标部分升高,符合HA20的表现,通过临床特征及基因检测联合确诊。治疗上先用免疫抑制剂,因药物副作用后启用生物制剂抗TNF-α制剂(英夫利西单抗),目前治疗方案可有效控制症状,需进一步随访病情变化及预后。
所有作者均声明本研究不存在利益冲突

























