
患儿,女,9个月,因"发热2 d,嗜睡、阵咳1 d,呼吸费力半天"就诊。
患儿既往生长发育正常,发病前1周添加配方奶粉,本次病程中出现低血糖、严重酸中毒、低血压表现。头颅MRI:双侧小脑半球、双侧基底节区见斑片状及小斑片状对称分布异常信号影,边缘不清,呈T1W1低信号、T2W1及黑水序列呈高信号。
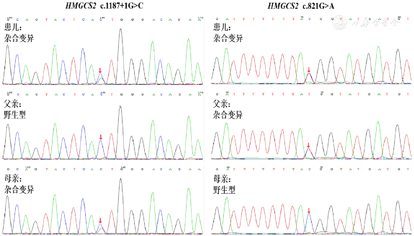
血乙酰肉碱升高,尿多种有机酸(3-羟基丙酸、3-羟基异戊酸、甘油酸、己二酸、辛二酸、3-羟基丁酸、3-羟基-4-己烯酸、5-羟基己酸、顺式-5-羟基-2-己烯酸、反式-3-羟基-4-己烯酸)增高,提示脂肪酸代谢障碍、多种羧化酶缺乏症、有机酸血症。HMGCS2基因(NM_005518)存在2个杂合变异,分别为4号外显子c.821G>A(p.R274H)杂合变异和6号内含子c.1187+1G>C杂合变异。
给予高碳水化合物、低蛋白、低脂肪饮食,补充肉碱,避免长时间空腹、超负荷运动、感染等应激状态。
患儿出院半年及1年随访,生长发育正常。
儿科;神经内科;遗传代谢科;重症医学科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
线粒体3-羟基-3-甲基戊二酰辅酶A合成酶缺乏症(HMG-CoA synthase deficiency,HMCSD)是一种罕见的常染色体隐性遗传病,病因是由于HMG-CoA合成酶缺乏导致催化乙酰辅酶A及乙酰辅酶A合成HMG-CoA及酮体生成障碍。临床主要表现为低酮症性低血糖症,代谢性酸中毒,嗜睡,脑病和肝肿大。在感染或禁食等应激状态下,急性发作,出现严重的低酮症性低血糖,严重的代谢和乳酸性酸中毒,严重者出现昏迷甚至猝死[1,2,3]。本文回顾分析重症监护室收治的1例HMCSD患儿的临床资料,并复习相关文献,以提高儿科医师对该病的认识。
患儿,女,9个月,因"发热2 d,嗜睡、阵咳1 d,呼吸费力半天"于2020年7月收入医院PICU。
病史:入院前2 d患儿发热,热峰38.6℃,无咳喘、吐泻等,至当地医院就诊,诊断上呼吸道感染,给予口服药物。入院前1 d患儿嗜睡、纳差、伴阵发性咳嗽,未行诊治,半天前患儿精神萎靡、反应差、呼吸费力,至当地医院诊断急性呼吸衰竭、休克,给予吸氧、生理盐水扩容后,转至我院PICU。患儿系第1胎第1产,母孕期体健,孕40周剖宫产,出生体重3.9 kg,身长50 cm,Apgar评分10分。生后母乳喂养,发病前1周添加配方乳及少量辅食,奶量减少。目前生长发育同正常同龄儿。家族史:父母体健,非近亲婚配,否认家族性遗传病史。
体格检查:体温37.5℃,脉搏175次/min,呼吸50次/min,血压71/37 mmHg(1 mmHg=0.133 kPa)。发育正常,营养中等,精神萎靡,反应差,鼻翼煽动,口周发绀,呼吸促,三凹征阳性,双肺听诊呼吸音粗,可闻及少量粗湿啰音。腹稍胀,肝肋下4.5 cm,质硬,脾脏肋下未触及,四肢肌力肌张力正常,毛细血管再充盈时间4 s。
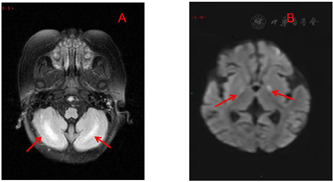
血气分析:pH 6.79,二氧化碳分压15.0 mmHg,氧分压181 mmHg,碳酸氢根2.1 mmol/L,碱剩余-28.9 mmol/L,乳酸0.4 mmol/L,阴离子间隙34.3 mmol/L,钾5.26 mmol/L,钠149 mmol/L,血糖2.1 mmo/L。肝功能未见明显异常。心肌酶及心脏彩超未见异常。头颅MRI示:双侧小脑半球、双侧基底节区见斑片状及小斑片状对称分布异常信号影,边缘不清(图1)。
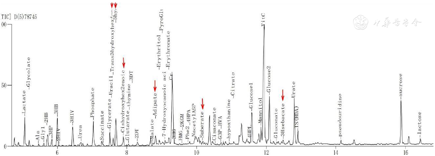
血尿遗传代谢筛查:血遗传代谢病筛查示,乙酰肉碱升高,余未见显著异常。尿气相色谱质谱显示,多种有机酸(3-羟基丙酸、3-羟基异戊酸、甘油酸、己二酸、辛二酸、3-羟基丁酸、3-羟基-4-己烯酸、5-羟基己酸、顺式-5-羟基-2-己烯酸、反式-3-羟基-4-己烯酸)增高(图2)。
基因检测:患儿HMGCS2基因(NM_005518)存在2个杂合变异,分别为4号外显子c.821G>A(p.R274H)杂合变异和6号内含子c.1187+1G>C杂合变异。采用PCR-Sanger测序方法进行家系验证,证实这两个变异分别遗传自父亲和母亲,为复合杂合变异(图3)。
本例患儿急性发作期,尿有机酸分析显示多种有机酸增高(3-羟基丙酸、3-羟基异戊酸、甘油酸、己二酸、辛二酸、3-羟基丁酸、3-羟基-4-己烯酸、5-羟基己酸、顺式-5-羟基-2-己烯酸、反式-3-羟基-4-己烯酸);恢复期尿有机酸分析,除了HMCSD相关有机酸指标异常(3-羟基-4-己烯酸、5-羟基己酸、顺式-5-羟基-2-己烯酸、3-羟基丁酸偏高),其余尿有机酸分析异常指标恢复至正常范围。首先怀疑患者体内存在脂肪酸氧化紊乱,酮体合成或利用缺陷,肝功能受损、有机酸血症、糖异生缺陷、线粒体疾病。结合血浆低浓度的酮体和正常酰基肉碱结果,鉴别诊断排除有机酸血症,糖异生缺陷和酮体利用缺陷,高度怀疑HMCSD。结合患儿基因检测结果HMGCS2基因(NM_005518)存在2个杂合变异,分别为4号外显子c.821G>A(p.R274H)杂合突变和6号内含子c.1187+1G>C杂合突变。其中,已有国内文献报道c.1187+1G>C变异[6],为剪接突变,推测将介导mRNA剪切时第6号外显子的跳跃,产生截断蛋白,为可能致病性变异,为非常强致病证据PVS1。该变异在公共人群数据库(千人基因组数据库、EXAC数据库、ESP数据库)中频率<0.0005,为中等致病证据PM2。患儿临床表现、生化检测以及血尿代谢物筛查高度符合HMCSD,支持致病证据PP4。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南,c.1187+1G>C判定为致病性变异(PVS1+PM2+PP4)。c.821G>A变异使得第274位氨基酸由精氨酸变异为组氨酸(p.Arg274His)。该变异在公共人群数据库(千人基因组数据库、EXAC数据库、ESP数据库)中频率<0.0005,为中等致病证据PM2。在隐性遗传病中,在反式位置上检测到致病变异,中等致病证据PM3。氨基酸变异危害性评估预测软件Polyphen2、SIFT和Mutationtaster预测该变异对基因产物有害,支持致病证据PP3。患儿临床表现、生化检测以及血尿代谢物筛查高度符合HMCSD,支持致病证据PP4。根据ACMG指南,c.821G>A判定为可能致病性变异(PM2+PM3+PP3+PP4)。采用PCR-Sanger测序方法进行家系验证,证实这两个变异分别遗传自父亲和母亲,为复合杂合突变。符合HMCSD表现。
入院后给予抗感染、机械通气、纠酸补碱、扩容补液以及纠正低血糖,维持内环境稳定等治疗;并予血液净化、输注悬浮红细胞等一系列治疗后,临床好转出院。
出院后以饮食控制为主,给予高碳水化合物、低蛋白、低脂肪饮食,补充肉碱,避免长时间空腹、超负荷运动、感染等应激状态。出院后半年、1年随访患儿生长发育正常。
HMG-CoA合成酶分为胞质型(HMGCS1)和线粒体型(HMGCS2),胞质内表达的HMGCS1主要与胆固醇合成有关,线粒体基质中表达的HMGCS2是酮体合成的限速酶[4]。线粒体HMCSD是一种罕见的酮体生成障碍性代谢紊乱,已报道的病例发病年龄多在3个月~6岁,主要表现包括呕吐、腹泻、肌张力低下、低体温、嗜睡、呼吸暂停甚至昏迷[5,6,7,8,9]。该病易被误诊,患者在无饥饿、感染等诱发之前通常是无症状的;在长时间禁食、感染或者腹泻后,患者迅速出现低血糖、代谢性酸中毒、心肌损害,被误诊为败血症、肾小管酸中毒、扩张性心肌病或心律失常等[2,3,4]。本报道患儿发病前1周添加配方乳及少量辅食,奶量减少,本次感染后出现低血糖、严重酸中毒、休克等,但乳酸正常,且阴离子间隙明显升高,高达34.3 mmol/L,与单纯脓毒性休克表现不符,因此考虑遗传代谢病的可能,奶量减少和感染可能是导致患儿出现低血糖等一系列临床症状的诱发因素。HMCSD患儿由于低血糖及异常代谢产物堆积造成细胞渗透压增高,神经细胞低灌注、神经元受损脑细胞受损,头颅MRI可出现双侧基底节区异常信号以及双侧额、颞、岛叶脑萎缩;多灶性脑白质异常信号和基底神经节损害是该病常见的神经影像学异常[11,12],与本例患儿头颅磁共振表现相符。
HMCSD患儿发病期通常存在低血糖、低酮体、代谢性酸中毒、转氨酶升高,血浆脂肪酸增高;急性发作期的尿气相色谱质谱检测常提示双羧酸增高,在经补充葡萄糖治疗后或疾病稳定期通常无显著异常[13]。虽然血浆酰基肉碱和尿液有机酸分析,对于脂肪酸氧化紊乱和HMCSD诊断不具有特异性[14],但在急性发作期的实验室检查结果(低酮症、游离脂肪酸升高、正常酰基肉碱、特定的有机酸尿症)具有很高的提示价值[13]。本例患儿急性发作期,尿有机酸分析显示多种有机酸增高(3-羟基丙酸、3-羟基异戊酸、甘油酸、己二酸、辛二酸、3-羟基丁酸、3 -羟基-4-己烯酸、5-羟基己酸、顺式-5-羟基-2-己烯酸、反式-3-羟基-4-己烯酸);恢复期尿有机酸分析,除了HMCSD相关有机酸指标异常(3-羟基-4-己烯酸、5-羟基己酸、顺式-5-羟基-2-己烯酸、3-羟基丁酸偏高),其余尿有机酸分析异常指标恢复至正常范围。患儿临床和尿有机酸分析结果使我们首先怀疑患者体内存在脂肪酸氧化紊乱,酮体合成或利用缺陷,肝功能受损、有机酸血症、糖异生缺陷、线粒体疾病。结合血浆低浓度的酮体和正常酰基肉碱结果,鉴别诊断排除有机酸血症,糖异生缺陷和酮体利用缺陷,高度怀疑HMCSD。此外,Pitt等[15]和Conboy等[16]研究发现,4-羟基-6-甲基-2-吡喃酮(4-HMP)增高,是HMCSD的潜在生物学标志物,具有强烈提示意义。本例中患儿2次尿筛均未检测到4-HMP的异常变化。文献报道急性期尿液4-HMP水平未见异常[7,17],可能与基因突变类型或标本留取时机有关。
HMGCS2基因主要在肝脏中高表达,在结肠、肾脏、睾丸、胰腺中低表达,在大脑、皮肤、外周血中不表达。HMG-CoA合成酶催化CoA和乙酰CoA合成HMG-CoA,该反应为酮体生成过程中的关键步骤。HMCSD的致病基因HMGCS2位于1号染色体p12-p13区域,编码10个外显子,含21 708个碱基对。目前国内外已报道的HMGCS2基因突变共57个。其中大多数是错义突变(37/57),无义突变8个,经典剪切突变5个。目前尚无基因型与表型相关性的可靠结论[18],但是也有研究认为基因突变所致的HMGS缺陷程度不同,HMCSD临床表现差异大[19]。本例患儿携带的两个杂合突变,分别是经典内含子剪切突变和错义突变。其中,c.1187+1G>C突变,已有国内文献报道[6],推测将介导mRNA剪切时第6号外显子的跳跃,产生截断蛋白,为可能致病性变异。c.821G>A突变使得第274位氨基酸由精氨酸突变为组氨酸,可能改变了蛋白结构、影响酶的功能,但尚需功能实验才能明确致病性。综合患儿的临床特点和基因检测结果,支持HMCSD。
HMCSD急性发作期的治疗包括纠正低血糖,代谢性酸中毒和促进有机酸代谢产物排泄。长期治疗的原则主要是预防低血糖发作,避免长时间空腹,随身携带食物剧烈运动后及时补充糖分[6,17]。本例患儿半年以及1年随访,患儿未再出现低血糖、酸中毒等情况,生长发育正常。
综上,本文对1例重症的HMCSD患儿救治的全过程进行报道,回顾分析了HMCSD的发病特点和遗传突变特点,对于临床上出现不明原因低血糖,尤其是低酮症性低血糖,以及严重酸中毒和循环障碍的患儿,要警惕罹患本病的可能。需尽早行相应的生化检测及代谢物筛查,及时进行有效的对症支持治疗,帮助患儿渡过疾病危险期。在疾病的恢复期,给予高碳水化合物,低蛋白、低脂肪饮食,可明显改善预后。

























