
患者,女性,47岁,因"步态不稳1年余,左上肢笨拙半年"入院。患者于2019年5月无明显诱因出现步态不稳,伴转身时头晕,外院考虑"多巴胺反应性肌张力障碍",予多巴丝肼口服未见好转。后步态不稳逐渐加重,2020年2月出现左上肢笨拙,精细动作变差,外院考虑"帕金森综合征",予普拉克索、丁苯酞治疗,症状无改善,存在步态不稳家族史。
神经系统体格检查:眼球扫视、追踪不平滑,上视欠充分,偶可见水平眼震,右侧掌颌反射(+),双侧轮替试验稍笨拙,双侧跟膝胫欠稳准,步基稍宽,似醉酒步态,直线行走不能。
外周血基因测序提示位于20号染色体上的PRNP基因存在错义突变,c.305C>T,综合临床表现、家族史及基因测序结果确诊Gerstmann-Sträussler-Scheinker综合征。
弥可保、复合维生素B、维生素B1营养神经,艾地苯醌改善脑代谢、金纳多改善脑循环。
头晕明显减轻,步态不稳稍差,左手活动基本同前,认知功能未见明显下降。
神经内科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
格斯特曼-施特劳斯勒尔-沙因克尔综合征(Gerstmann-Straussler-Scheinker syndrome,GSS综合征)是一种非常罕见的朊蛋白病,临床主要表现为小脑性共济失调,逐渐进展,可伴不同程度的痴呆。本病较为罕见,发病率为每年每1亿人中发病1~10例[1]。GSS综合征呈常染色体显性遗传,几乎完全外显。在世界范围内已发现了24个独立的GSS综合征家族。本文报道1例经基因检测确诊GSS综合征的患者,并对文献进行复习以期为GSS综合征的诊断提供更多临床参考。
患者,女性,47岁。因"步态不稳1年余,左上肢笨拙半年"于2020年11月2日入院。患者于2019年5月无明显诱因逐渐出现步态不稳,表现为站立及行走时身体摇晃,缓慢步行可、上楼可,下楼费力,伴转身时头晕,无双下肢无力、麻木及踩棉感,无恶心、呕吐、天旋地转感,无黑矇、晕厥。2019年9月就诊于外院,查颈椎MRI、头MRI未见明显异常;躯体感觉诱发电位、脑干听觉诱发电位、视觉诱发电位及肌电图检查未见明显异常,考虑"多巴胺反应性肌张力障碍?",予多巴丝肼3/4片、3次/d口服,效果欠佳。其后步态不稳症状逐渐加重。2020年2月患者出现左上肢笨拙,表现为较对侧活动不灵活,系扣、写字等精细动作变差。2020年8月患者出现睡眠中肢体抽动样动作,2~3次/月,就诊于外院。肌电图:F波,左侧正中神经出现率下降,双侧腓总神经未引出,余正常。头MRI、脑电图、多导睡眠图未见明显异常,考虑"帕金森综合征",予普拉克索、丁苯酞软胶囊治疗,症状无改善。既往高血压、高脂血症病史,家族史:母亲60岁左右出现步态不稳,后逐渐加重,但仍能行走,72岁时因心梗去世。姐姐63岁诊断"朊蛋白病可能",以步态不稳起病,后逐渐痴呆,基因检测示PRNP基因c304C>T杂合突变,导致氨基酸改变p.P102L(脯氨酸>亮氨酸),1年后病逝,余3个哥哥及1个姐姐无相似表现。其他无特殊。
神经系统体格检查:神清语利,高级智能可,眼球扫视、追踪不平滑,上视欠充分,偶可见水平眼震,余颅神经(-),四肢肌力、肌张力可,腱反射对称引出,双侧Babinski征、Chaddock征(-),右侧掌颌反射(+),深浅感觉正常,双侧轮替试验稍笨拙,双侧跟膝胫欠稳准,Romberg征(-),步基稍宽,似醉酒步态,直线行走不能,足跟、足尖行走可,脑膜刺激征(-)。简易精神状态检查评分30分(初中文化水平),蒙特利尔认知评估量表评分25分。
实验室检查:血常规、便常规、血糖、甲状腺功能、红细胞沉降率、超敏C反应蛋白等检验结果均在正常参考值范围内。尿常规+沉渣:白细胞计数500 Cells/μl,PRO TRACE g/L,余正常。肝肾功能:K 3.4 mmol/L,余正常。血脂:三酰甘油3.68 mmol/L,余正常。抗谷氨酸脱羧酶抗体、ANCA(-);ANA抗体谱:ANA(+)核点型1∶80,AHA弱阳性(+)18,余阴性。
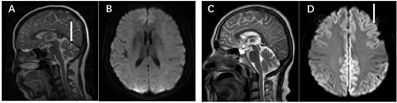
影像学检查:头常规MRI+SWI示,小脑欠饱满,余未见明显异常(图1)。经颅多普勒超声示,各血管血流频谱未见明显异常。
脑电图检查结果:未见明显异常。
脑脊液检测:腰穿压力170 mmH2O(1 mmH2O=0.0098 kPa),脑脊液常规、生化、细胞学、寡克隆区带分析、抗神经抗原抗体检测(Ri+Hu+Yo)、细菌涂片+培养、抗酸染色、真菌涂片均阴性。
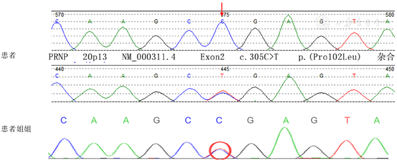
外周血基因测序(Sanger):在20号染色体的PRNP基因外显子区域发现一处杂合突变:c.305C>T(胞嘧啶>胸腺嘧啶),导致氨基酸改变:p.P102L(脯氨酸>亮氨酸)(杂合变异,箭头及红圈所指)(图2)。家系验证结果显示其姐姐同样携带该位点的杂合变异,其母亲虽有相似共济失调表现,但现已去世,未做基因检测。该变异在dbSNP147数据库有收录,ESP6500、千人基因组数据库均未见收录,生物信息学软件预测其有治病可能性,根据美国医学遗传学与基因组学会发布的变异解读指南分析为致病型(强致病证据PS1+PS3+PS4)。
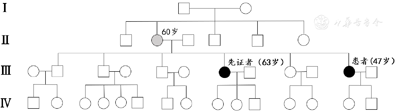
家系验证结果:其姐姐以步态不稳起病,首先证实携带相应位点的杂合变异,其母亲存在步态不稳病史,但未行基因检测,余3个哥哥及1个姐姐均体健,家系图见图3。
根据临床表现、阳性家族史及基因检测结果确诊GSS综合征综合征。
予弥可保、复合维生素B、维生素B1营养神经,艾地苯醌改善脑代谢、金纳多改善脑循环。
患者症状稳定,2020年11月19日出院。电话随访(2年后):头晕明显减轻,步态不稳稍差,左手活动基本同前,认知功能未见明显下降。
朊蛋白病是一种罕见的神经退行性疾病,预后不良,其特征表现为正常的细胞朊粒蛋白通过错误折叠形成富有β折叠结构的构象异构体,即羊瘙痒病朊粒蛋白。根据病因的不同,分为散发性、获得性及遗传性三种类型。其中散发性朊蛋白病是最常见的类型,占85%~90%,主要包括散发性克-雅病(Creutzfeldt-Jakob disease,CJD)及另外两种更为少见的疾病:变异型蛋白酶敏感朊蛋白病和散发性致死性失眠症。获得性朊蛋白病最为罕见,占比不到1%,包括库鲁病、医源性CJD以及变异性CJD。遗传性朊蛋白病占10%~15%,是由于PRNP(编码PrP的基因)发生常染色体显性突变引起,共有三种临床表型,分别为家族性CJD、GSS综合征、致死性家族失眠症。
GSS综合征由位于20号染色体的PRNP基因的不同错义突变引起,几乎完全外显,其中最常见类型的是P102L突变(第102位氨基酸残基由脯氨酸变为亮氨酸)。1913年,GSS综合征的首例家系由一位维也纳神经精神专家发现并报道,迄今为止,至少发现16种不同的错义突变类型与其发病相关,包括P84S、P102L、P105L、P105S、A117V、G131V、S132I、V176G、H187R、F198S、D202N、E211D、Q212P、Q217R、Y218N和M232T[2]。
GSS综合征临床主要表现为小脑性共济失调、认知功能下降、言语和吞咽障碍、感觉障碍、锥体外系损害及帕金森症状,小脑病变表现包括动作笨拙、不协调和步态共济失调。在印第安纳州的家系中,GSS综合征患者特殊存在眼球运动异常,包括凝视诱发眼球震颤、回弹性眼球震颤、顺畅追赶受损和高眼球跳动等。最近Nicole报道了1例核上性眼肌麻痹的GSS综合征患者,其眼球运动与本患者表现极为相似,均表现为眼动追随不畅、上视不充分,不同的是该患者存在F198S基因突变[3]。GSS综合征平均病程49个月,但波动范围较大,从7个月到132个月不等,70%的患者有家族史[4,5]。GSS综合征的临床表现、病情进展存在较大的家族及个体差异,部分程度上可能是由潜在PRNP基因突变差异或密码子129相关多态性的差异所致。例如存在P102L突变的GSS综合征患者,小脑症状可能更显著[6]。而存在A117V和M232T突变的患者,痴呆可能更显著,Y145STOP患者则主要表现为痉挛性截瘫及进展性痴呆[1,7]。在所有GSS综合征患者中,均可以根据129号密码子的多态性分为蛋氨酸纯合子和蛋氨酸/缬氨酸杂合子两种类型。研究指出,在P102L突变类型患者中,蛋氨酸纯合子患者发病年龄较蛋氨酸/缬氨酸杂合子患者大约提前7岁[4]。
GSS综合征无法通过实验室或影像学检查做出诊断,GSS综合征的脑脊液结果往往正常,脑电图可显示慢波。早期阶段,头MRI常无明显异常发现,后可表现为非特异性大脑皮层或小脑萎缩,单光子发射计算机断层扫描可显示脑血流弥漫性减少,呈马赛克状,尤其是枕叶及脊髓,而小脑的灌注却相对保留[8]。PRNP基因检测相对具有更高的敏感性及特异性,迄今调查的所有GSS综合征家族均存在PRNP基因突变。
不同于CJD典型的大脑皮层海绵状改变,GSS综合征的神经病理学特征性表现为多中心PrP-淀粉样蛋白斑块沉积,伴放射状针状体,羊瘙痒病朊粒蛋白免疫染色及糖原染色呈阳性,主要分布于小脑分子层,在一些患者中,这些斑块可呈光晕表现[5]。通过超微结构分析,淀粉样沉积物是由平均17.64 nm的纤维组成的致密网状结构[9]。在密码子105、145及217突变的患者中,可观察到显著的tau蛋白阳性的神经纤维缠结,在Western blot中,GSS综合征患者和PRNP突变携带者通常可检测出分子量7~10 kD的抗蛋白酶K分解的羊瘙痒病朊粒蛋白片段[5]。
综上,本例患者以小脑性共济失调、精细动作困难、眼动异常为突出临床表现,未出现典型认知功能减退症状,外院曾误诊为帕金森相关疾病,朊蛋白病阳性家族史在疾病的诊断过程中发挥着关键作用,指导我们在临床工作中重视对家族史的询问及加强眼球运动异常表现的思考。对于临床中高度怀疑朊蛋白病的患者,要重视PRNP基因的检测,以减少临床中对此病的误诊或漏诊。
所有作者均声明不存在利益冲突

























