
学龄前期男童,因"反复低钠血症并抽搐"就诊。实验室检查提示低血钠、低血浆渗透压、高尿钠、高尿渗透压,二代测序示患儿AVPR2基因存在c.409C>T(R137C)变异,最终确诊。
反复抽搐,生长发育同同龄儿。
结合患儿为幼儿,反复低钠血症、抽搐,补钠效差,查血容量正常,渗透压降低,尿钠明显增多,临床和基因均明确为肾性抗利尿不当综合征。
预限水治疗(1000 ml/m2),血钠可维持在正常范围,出院后给予限水(1200 ml/m2)+氯化钠0.5 g。
每周复查血钠可以维持134~139 mmol/L之间,未再出现抽搐。
儿科;内分泌遗传代谢科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
低钠血症在临床中为很常见的电解质紊乱,其中抗利尿激素不适当分泌为其病因之一,使水排泄受阻,导致低钠血症(<135 mmol/L),低血浆渗透压(<275 mmol/L),尿浓度过高(>100 mOsm/kg),尿钠排泄(>40 mmol/L)。其常见的病因包括恶性肿瘤、原发性疾病、神经系统疾病、各种药物及其他如疼痛,恶心和急性间歇性卟啉症,以上病因是由于抗利尿激素分泌增多,激活精氨酸加压素受体2(AVPR2),导致肾小管吸收水增多[1]。而由10%~20%的患者是由于AVPR2激活突变引起的肾性抗利尿不当综合征(nephrogenic syndrom of inappropriate antidiuresis,NSIAD)(OMIM 300539),是一种罕见的X连锁的隐性遗传病,由Feldman等[2]首次报道,NSIAD与抗利尿激素分泌不适当综合征不同的是患者的精氨酸加压素(AVP)无分泌异常增加,而是呈现低水平。AVPR2受体失活变异导致肾性尿崩症较为常见,激活变异引起的尿液浓缩,尿量减少,导致NSIAD罕见。迄今为止,国内仅有2篇报道[3,4]。早期诊断和治疗可以改善预后。现回顾分析本院内分泌遗传代谢科确诊的1例肾性抗利尿不当综合征患儿临床及遗传学特点,并通过文献复习,总结NSIAD的临床特征及基因变异特点,提升对其的认知和诊疗水平。
患儿,男,3岁1个月,间断抽搐并低钠血症1个月,于2022年5月4日就诊于本院内分泌遗传代谢科。1个月前患儿无明显诱因出现恶心、呕吐,相继出现抽搐,表现为双眼上翻、口周流涎、牙关紧闭、四肢强直伴抖动,呼之不应,伴小便失禁,无大便失禁等,持续约2 min自行缓解,缓解后困乏,无多饮、多尿,否认纳差、水肿,否认多汗,无口服特殊药物病史,急诊来我院诊治,查电解质示:血钠111.0 mmol/L,遂入我院神经内科住院诊治,诊断为"1.惊厥发作;2.胃肠功能紊乱;3.低钠血症;4.脑电图异常",给予补钠及维生素B6针及其他对症治疗,再未发生类似抽搐样症状,动态复查电解质,出院前血钠升高至125 mmol/L,未明确病因出院,出院后精神状态可,未给予补钠及监测电解质。半个月前疑似饮食不当(幼儿园过度饮食)出现恶心、呕吐,于当日再次发生同前抽搐样症状,伴意识不清,持续约3 min后自行缓解,再次急诊入我院神经内科诊治,入院时测血钠117.0 mmol/L,诊断为"1.胃肠功能紊乱;2.惊厥发作;3.低钠血症",继续予补钠及其他对症治疗,动态监测电解质,血钠升高至125 mmol/L后出院治疗。出院至此次入院前口服补液盐[每次5.125 g(相当于氯化钠0.85 g)。每天3次]治疗,入院2 d前我院复查血钠为122.6 mmol/L,至外院复查血钠仍提示血钠低,波动于123 mmol/L。1年前因"高热惊厥"在我院神经内科住院诊治,当时测血电解质正常,血钠135 mmol/L。患儿系其母第二胎第二产,足月顺产,BW 3.05 kg,生长发育及喂养史无特殊,平素喜食咸食,不喜饮水,每日饮水量900 ml左右,尿量500 ml左右。父母体健,有1个哥哥,5岁,体健,否认家族中类似疾病史及遗传病。
入院体格检查:血压92/58 mmHg(1 mmHg=0.133 kPa),身材匀称,身高100.2 cm(75 th~90 th),体重14.5 kg(50 th~75 th),BMI 14.44 kg/m2,头围49 cm,神志清楚,精神反应好,全身皮肤无干燥,眼窝无凹陷,口唇红润,无干燥,咽无充血,颈软,无抵抗,心肺腹查体无异常,神经系统查体无异常。
血钠123 mmol/L时查血气分析无酸碱失衡,甲功正常,肾素-血管紧张素-醛固酮系统(卧位):醛固酮174.641 pg/ml(25~129 pg/ml),肾素8.199 pg/ml(4~24 pg/ml),血管紧张素Ⅱ 306.717 pg/ml(25~129 pg/ml);肾素-血管紧张素-醛固酮系统(立位):醛固酮994.622 pg/ml(40~310 pg/ml),肾素96.299 pg/ml(4~38 pg/ml),血管紧张素Ⅱ 323.876 pg/ml(49~252 pg/ml);0点、8点、16点促肾上腺皮质激素分别为14.9、36.43(7.2~63.3)、28.09 pg/ml,0点、8点、16点皮质醇3.91、7.16(7.26~32.28)、6.52(3.24~15.00)μg/dl,17α-羟基孕酮1.112 ng/ml(0.31~2.01 ng/ml),肾功能示:尿素2.4 mmol/L(2.7~7.0 mmol/L),肌酐21.2 μmol/L(19~44 μmol/L),尿电解质示:尿量0.51 L,尿钙4.14 mmol/L,24 h尿钾13.24 mmol/24 h(25~100 mmol/24 h),24 h尿钠65.18 mmol/24 h(130~260 mmol/24 h),24 h尿氯50.54 mmol/24 h(170~250 mmol/24 h)。入我科后查电解质示:血钠128 mmol/L时尿素3.7 mmol/L,肌酐22.8 μmol/L,尿钠164.64 mmol/24 h,尿比重1.030,完善血钾、血钙、血糖、AFP、CEA、HCG等肿瘤标志物未见异常,完善胸部、腹部CT、头颅和垂体核磁共振成像未见占位性病变,完善视频脑电图未见明显异常,韦氏智力测试量表测试98分,提示智力正常。父母及哥哥血电解质均正常,随机尿尿比重1.015。
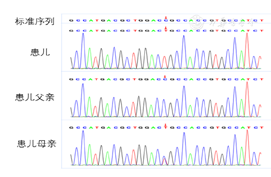
基因检测(图1);患儿反复低钠,临床考虑肾性抗利尿不适当综合征,行全外显子基因检测协助诊断。获得患儿父母知情同意并签署书面同意书后,且经医院伦理审查委员会批准,抽取患儿及父母、哥哥外周血各2 ml,送北京智因东方转化医学研究中心进行检验。患儿行全外显子测序,并Sanger法测序验证,参考基因组(hg19)发现患儿X染色体上AVPR2基因存在编码区第3外显子c.409C>T(p.R137C,p.Arg137Cys)(NM_000054),半合子变异,来源于母亲,父亲为野生型,哥哥为野生型。参考美国医学遗传学和基因组学协会(American College of Medical Genetics and Genomics,ACMG)基因变异解读指南,c.409C>T为罕见变异,在dbSNP、千人基因组、EXAC、gnomAD、gnomAD东亚数据库中没有收录,且为HGMD数据库已报道的致病变异,导致137位半胱氨酸代替精氨酸,评级为致病变异(PM5-Strong+PS3+PM1+PM2+PP3)。
根据实验室检测结果,患儿无高血糖、应用甘露醇药物,因此高渗性低钠血症可排除;无高脂血症、无高蛋白血症,故等渗性假性低钠血症不考虑;尿钠浓度反复增高>30 mmol/L,提示为正常血容量性低钠血症,常见原因有糖皮质激素缺乏、腺垂体功能减退、甲状腺功能减退、大量摄入水或低钠液体、SIADH,该患儿皮质醇节律、甲状腺功能均正常,无大量摄入低张液体病史,故考虑SIADH。患儿年龄小,无感染、肿瘤、特殊药物服用史,结合基因变异分析,NSIAD诊断明确。
入院后给予仅生活方式干预限水治疗(1000 ml/m2),后因家长自觉患儿每日入水量少,调整为限水(1200 ml/m2)+氯化钠0.5 g。
入院后给予仅生活方式干预限水治疗(1000 ml/m2),血钠可维持在正常范围,出院后给予限水(1200 ml/m2)+氯化钠0.5 g,每周复查血钠可以维持134~139 mmol/L之间,未再出现抽搐,随访6个月生长发育正常。
低钠血症在临床中为很常见的电解质紊乱,常继发于消化道、肾上腺、肾脏及心脏等基础疾病,有些病因不明,大多数给与补钠后可升至正常。该患儿反复低钠血症,有惊厥病史,低钠血症导致细胞外液及循环血容量增加,促进脑极夜生成,使颅内压增高,从而产生惊厥等神经症状,虽然完善头颅CT无脑水肿发生,但患儿惊厥考虑与电解质紊乱或轻度脑水肿有关。患儿低钠血症给与补充钠盐效差,故反复低钠血症的患儿应考虑血容量及血浆渗透压,血浆渗透压正常或升高的患儿应排除假性低钠血症。血浆渗透压降低者进一步评估血容量,不同血容量状况低钠血症的主要病因不同,该患儿血浆渗透压降低,血容量正常,常见原因有糖皮质激素缺乏、腺垂体功能减退、甲状腺功能减退、大量摄入水或低钠液体、SIADH,该患儿皮质醇节律、甲状腺功能均正常,无大量摄入低张液体病史,故考虑SIADH。患儿年龄小,无感染、肿瘤、特殊药物服用史,完善基因检测提示存在AVPR2基因变异,故诊断NSIAD。
NSIAD是肾性尿崩症(NDI)的镜像。在NDI中,肾脏不能浓缩尿液,而在NSIAD中,尿液稀释受损,与抗利尿激素的存在与否无关。因此,NDI患者有高钠血症脱水的风险,而低钠血症是NSIAD的典型表现,类似于不适当的抗利尿综合征[26]。
AVPR2基因位于Xq28,编码VAP受体蛋白,一种G蛋白偶联受体。在肾小管远端基底膜外侧表达,通过激活G蛋白增加细胞内cAMP,减少尿液产生,参与和维持水的稳态[27]。AVPR2基因包括2.2 kb跨膜区和3个外显子。目前国内外报道的NSIAD已60余例,最常见的AVPR2变异为错义突变,其中R137C和R137L导致构成性β-抑制素募集和受体内吞,破坏细胞表面靶向,降低细胞表面表达,因此,这些活性受体对抗利尿激素刺激和受体抑制剂无反应。相反,其他AVPR2变异类型F229V,I130N和L312S对受体抑制剂有反应[13,18,20]。中国共报道NSIAD病例5例,均为R137C变异,提示R137C为中国常见变异。
由于NSIAD罕见,其患病率尚不明确。主要症状是低钠血症,大多数婴儿及儿童以惊厥、呕吐起病,在男性患者中,大多数AVPR2基因突变是有低钠血症症状的,其中只有1例无低钠血症[12]。低钠血症的严重程度与临床表现无明显相关性,2例患儿生后即有惊厥症状,但有的患者到成年期发病,有的自始至终无低钠血症的症状,可能大脑适应了患者长期慢性低钠[5,7,14,18,19,23]。在女性患者中,大部分无低钠血症,其中10例存在低钠血症,4例在水负荷实验后出现尿少低钠血症,提示虽然女性为携带者,但仍有53.8%的女性患者,可能与X染色体随机失活有关。我们患者的母亲同样携带致病变异,但无低钠血症相关症状,查电解质正常。患儿查24 h尿钙增高,可能与患儿长期补钙有关,也可能患儿低钠血症,引起盐皮质激素增多,引起肾小管重吸收钙,引起的高尿钙。
对大部分患者限水治疗可使血钠恢复正常,部分患者需要加用钠盐、尿素或呋塞米治疗,托伐普坦可用于特异性基因变异(F229V、I130N和L312S)。我们的患者考虑NSIAD后即给予限水治疗,可使血钠升至正常,但由于母亲担心孩子摄入液体量太少,故我们适当加大液体摄入量同时给予氯化钠治疗,血钠维持良好,多次复查均在正常范围,未再出现低钠血症相关的症状。国内外报道的R137C变异为常见变异的患者发病年龄不一,有些患儿婴幼儿期既有惊厥、低肌张力、易激惹、呕吐,有些患者儿童期无症状直到成人期仅有轻度的恶心、呕吐、头痛症状,成人期起病的患者大部分仅需限水即可使血钠恢复正常,长期随访无不适。婴幼儿起病的患儿一般需要限水+补充钠,甚至需要口服尿素治疗,长期随访症状明显好转,无惊厥等症状发生。
因低钠血症在临床中是常见的,病因繁多,而NSIAD相对罕见,容易误诊和漏诊,长期低钠血症容易引起神经系统损害,故及时诊断和纠正低钠血症是必要的。故如遇长期不易纠正的低钠血症或有家族史的患者,尤其婴幼儿,及早行AVPR2基因检测,早期诊断和治疗。
所有作者均声明本研究不存在利益冲突

























