
患者,女性,45岁。因"发作性右侧肢体无力2年余,加重1周"入院。既往"右眼视盘血管炎"病史,甲泼尼龙治疗后好转。
以发作性右侧肢体无力、口齿不清起病,最后一次发作查体右下肢肌力3级,四肢腱反射亢进,双侧踝阵挛阳性,双侧病理征(+)。
根据2017 McDonald修订标准及参考文献对高活动性多发性硬化的定义。2年内临床发作4次(≥2次/年);MRI在脑室周围、幕下、颈髓3个区域,每个区域≥1个T2病灶,同时存在增强与非增强病灶;脑脊液寡克隆区带阳性(2型)。诊断:高活动性多发性硬化(highly active multiple sclerosis,HAMS)。
急性期予甲泼尼龙冲击、丙种球蛋白调节免疫,并加用DMT药物硫唑嘌呤序贯治疗,症状有所缓解,但仍反复复发加重,诊断高活动性多发性硬化后DMT药物调整为口服西尼莫德。
肢体无力症状基本完全缓解,随访至2022年8月,16个月未再复发,无残疾进展。
神经科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
多发性硬化(multiple sclerosis,MS)是一种免疫介导的中枢神经系统慢性炎性脱髓鞘性疾病,病变主要累及白质,其发作性症状表现各异,多见于疾病早期和病情恶化时,以发作性症状起病的患者易被误诊为中枢神经系统的其他疾病,如短暂性脑缺血发作、癫痫等,早期识别难度大。MS的自然病史具有高度异质性,高活动性多发性硬化(highly active multiple sclerosis,HAMS)具有早期、意外获得残疾,频繁复发(通常不完全缓解)以及磁共振(magnetic resonance imaging,MRI)上高活动性等特点。HAMS临床上少见,具有较高的早期进展风险,因此早期识别尤为重要,以"发作性肢体无力、口齿不清"起病的HAMS患者更为罕见,增加了早期正确识别的难度。现报道1例以"发作性肢体无力、口齿不清"为首发症状的HAMS,希望借此提高临床医生对本病的认识,争取早期识别,降低其致残率,提高患者生活质量。
患者,女性,45岁,以"发作性右侧肢体无力、口齿不清"起病,因"发作性右侧肢体无力2年余,加重1周"于2021年4月收住苏州大学附属第二医院。患者2019年2月无明显诱因出现发作性右侧肢体无力、口齿不清,右手持物不稳,右下肢行走拖曳,持续约1 min缓解,发作频率7~8次/d,无饮水呛咳、吞咽困难,无四肢抽搐、意识不清,外院就诊考虑短暂性脑缺血发作,予改善循环治疗无好转,上述症状仍反复发作,后未继续治疗。2020年4月症状较前加重,出现四肢乏力不缓解,行颅脑MRI提示脑室旁脑白质及脑干异常信号,未予治疗。2020年7月患者肢体无力较前进一步加重,行走时跌倒2次,至我院就诊,神经系统查体示:双下肢肌力4级,右侧指鼻试验欠稳准,四肢腱反射亢进,左侧踝阵挛阳性,双侧病理征(-),余无明显阳性体征。头颅、颈胸椎MRI(2020年7月):幕上及幕下脑白质内多发炎性脱髓鞘;C4-6椎体水平颈髓斑片状强化、脑桥及延髓内异常信号影,小脑内异常强化(图1)。脑脊液寡克隆区带阳性(2型)。结合患者病史、体征及影像学表现,依据2017版McDonald MS诊断标准[1],诊断"多发性硬化" 。扩充残疾功能量表(EDSS)评分:1.5分。予甲泼尼龙冲击,联合硫唑嘌呤免疫抑制治疗,出院时患者肢体无力症状减轻,双下肢肌力4+级。2020年11月复查颅脑、颈椎MRI较2020年4月对比,双侧脑室及四脑室旁白质、脑干病灶范围缩小,强化程度明显减低。2021年2月患者右下肢无力再发加重,行走不能,再次入住本院神经科。查体示:右下肢肌力3级,四肢腱反射亢进,双侧踝阵挛阳性,双侧病理征(+)。EDSS评分:3分。完善MRI较(2020年11月)相比:延髓及中脑左侧份新增小斑片状明显强化灶,C2椎体水平颈髓内新增斑片状强化灶;横断位示颈髓内多发斑片状强化灶,余较前相仿。实验室检测未见明显异常。腰椎穿刺压力175 mmH2O(1 mmH2O=0.0098kPa ),常规、生化正常范围。入院后予醋酸泼尼松小剂量口服,联合丙种球蛋白静滴,症状无明显改善,后加用甲泼尼龙500 mg冲击治疗,右下肢无力明显改善,出院时右下肢肌力4+级。患者病情反复,出院1周后右下肢无力再次加重,查体右下肢肌力3级,EDSS评分:2分。拟"高活动性多发性硬化"入住我科。病程中,患者神志清、精神一般,饮食可、睡眠欠佳,大小便正常,体重无明显变化。个人史及家族史无殊。
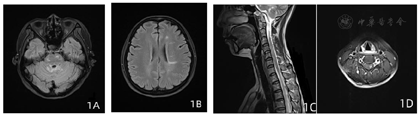
入院体检:神志清,对答切题,双侧瞳孔等大等圆,直径3 mm,光反应灵敏,眼球活动自如,未及眼震,伸舌居中,双上肢、左下肢肌力5级,右下肢肌力3级,四肢肌张力正常、痛觉对称存在,四肢腱反射亢进,双侧踝阵挛阳性,双侧病理征(+),双侧指鼻试验及左侧跟膝胫试验完成稳准。颈软,Kernig征(-)。EDSS评分:2分。
辅助检查:血尿粪常规、凝血系列、血生化全套、电解质、糖化血红蛋白、叶酸、维生素B12、肿瘤标志物、甲状腺功能、风湿因子、自身抗体初筛等未见明显异常。脑脊液寡克隆区带阳性(2型)。血、脑脊液AQP4抗体阴性。颅脑、颈胸椎MRI(平扫+增强):幕上及幕下脑白质内多发炎性脱髓鞘病灶;C4-6椎体水平颈髓斑片状强化、脑桥及延髓内异常信号影,小脑内异常强化。
本例患者以"发作性右侧肢体无力、口齿不清"起病,后反复发作,2年内发作4次(≥2次/年),激素冲击治疗有效,症状不完全恢复,磁共振成像可见幕上、幕下、脊髓多发脱髓鞘病灶,每次发作颅脑、脊髓MRI显示有新的病变或病变大小较前增加,脑脊液寡克隆区带阳性(2型),加用疾病修正治疗(disease-modifying treatment,DMT)药物西尼莫德后症状基本完全缓解、随访16个月未再发。诊断: HAMS。
该患者以反复发作性肢体无力、口齿不清起病,影像学呈现颅脑、脊髓脱髓鞘表现,根据患者疾病特点,需与以下疾病进行鉴别:
1.短暂性脑缺血发作(transient ischemic attack,TIA):多有"高血压、动脉粥样硬化、糖尿病、高血脂"等脑血管疾病危险因素,症状可表现为一过性口齿不清,失语,偏侧肢体无力或感觉障碍,眩晕伴恶心呕吐等,每次发作持续数分钟至数小时,可反复发作,但24 h内完全恢复,不遗留神经功能缺损,神经系统查体无明显定位体征,头颅影像学检查无责任病灶。本例患者以反复发作性肢体无力、口齿不清起病,早期与TIA难以鉴别,因患者无相关危险因素,后期完善脑脊液OB阳性、颅脑MRI+MRA示颅内多发脱髓鞘表现、颅内血管未见异常,故不支持该诊断。
2.癫痫:特别是单纯部分性发作,常表现为持续数秒至数分钟的肢体抽搐或麻木针刺感,从躯体的一处开始,并向周围扩展,脑电图可见尖波、棘波、尖-慢波或棘-慢波等痫样放电,CT/MRI检查可能发现脑内局灶性病变。本例患者表现为发作性右侧肢体无力、口齿不清,脑电图未见异常,MRI示颅内、颈髓多发脱髓鞘表现,故不支持该诊断。
3.视神经脊髓炎谱系病:是免疫介导的主要累及视神经和脊髓的原发性中枢神经系统炎性脱髓鞘疾病。其脑组织损害常累及抗水通道蛋白4(AQP-4)高表达区域,如下丘脑、邻近室管膜区(侧脑室、导水管周围、第三脑室旁、第四脑室旁)、胼胝体、脑干。脊髓受累长度≥3个节段, AQP4抗体表达阳性。本例患者无视神经受累表现,脊髓受累长度<2个节段,脑脊液寡克隆区带阳性,故不支持视神经脊髓炎的诊断。
患者于2020年7月行甲泼尼龙冲击500 mg、3 d,240 mg、3 d,120 mg、3 d,改为口服泼尼松60 mg,每周减5 mg,直至减停,同时联合硫唑嘌呤50 mg、1次/d免疫抑制治疗,肢体无力症状明显改善。于2021年2月症状加重,再次予甲泼尼龙冲击500 mg、5 d,240 mg、3 d,120 mg、3 d,80 mg、3 d,改为口服甲泼尼龙32 mg,每10天减4 mg,减至20 mg维持,同时联合丙种球蛋白静滴,症状再次改善。2021年3月症状再发加重,治疗上加用DMT药物西尼莫德2 mg、1次/d,症状明显缓解。
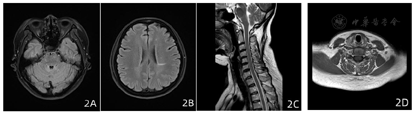
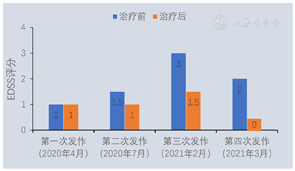
患者发作性右侧肢体无力,激素冲击治疗有效,症状不完全恢复,仍反复发作加重,自加用DMT药物西尼莫德,患者肢体无力症状基本完全缓解,自2021年4月出院后随访至2022年8月,16个月未再复发,末次复查颅脑、颈椎MRI(2021年9月)同2020年7月对比可见脱髓鞘病灶范围明显减小(图2)。现患者无不适主诉,双眼视力1.0,四肢肌力5级,双侧病理征(-),EDSS评分:0分,图3为患者治疗前后EDSS评分比较。
多发性硬化(MS)的发作性症状有三叉神经痛、癫痫、构音障碍、共济失调、复视、瘙痒、肢体感觉异常、肢体无力、强直性痉挛、Lhermitte'sign等,具有突发突止、短暂、刻板重复的特征[2]。其机制最流行的理论是由Osterman等[3]提出的,即部分脱髓鞘的轴突有利于自发神经冲动的异位产生,并横向传播到纤维束内的其他轴突,最终表现为阵发性发作。临床上以"发作性肢体无力、口齿不清"为首发症状的MS相对少见,误诊率高,本例患者以"发作性右侧肢体无力、口齿不清"起病,早期识别困难,临床上易考虑为中枢神经系统疾病,如TIA、癫痫等。希望通过对本例患者起病特点的认识,提高对以发作性症状起病MS的早期识别。对早期以发作性症状起病的患者,若颅脑、脊髓MRI无脑血管病相应特征,而出现脱髓鞘表现,应进行脑脊液寡克隆带检查,明确有无多发性硬化可能。
HAMS与其他MS不同,具有早期、意外获得残疾,频繁复发(通常不完全缓解)以及MRI上高活动性等特点,与反复严重发作和残疾加速累积相关,是"快速进展"的多发性硬化。MS广泛存在的神经轴突损伤和丢失可能被视为HAMS的神经病理学对应物。在临床实践中,尽管使用一个或多个DMT的治疗,持续的轴突丢失可能是持续进展患者的潜在病理[4]。
HAMS目前尚无统一定义,根据已有文献其被定义为具有以下一个或多个特征的RRMS表型:(1)EDSS评分在发病5年时为4分;(2)在1年中多次复发(≥2次)且症状不完全恢复;(3)2次以上的MRI研究显示T2中有新的病变或病变大小增加,或病变经钆处理后增强;(4)使用一种或多种DMT药物治疗至少1年没有反应。众多研究表明,MS患者前2年不受控制地复发加速早期疾病进展,而后期复发对疾病进展影响较小[5],因此早期识别HAMS尤为重要,认识快速转化为SPMS的预测因子[6],有助于临床医师对HAMS的早期识别:(1)短时间内EDSS评分累积3分;(2)在疾病发展过程中早期复发率高;(3)复发间隔时间短。
本例患者以"发作性右侧肢体无力、口齿不清"起病,后进行性加重,2年内反复4次(≥2次)发作,激素冲击治疗有效,症状不完全恢复,核磁共振可见幕上、幕下、脊髓多发脱髓鞘病灶,每次发作颅脑、脊髓MRI显示有新的病变或病变大小较前增加,脑脊液寡克隆区带阳性(2型),加用DMT药物西尼莫德后症状基本完全缓解、未再发。诊断:HAMS。
与其他MS患者不同,HAMS患者有很高的疾病进展风险,他们的治疗时间窗很窄,需要一个强大的诱导治疗而不是标准的免疫调节逐步升级疗法。因此,提高早期治疗的最佳疗效是必要的。诱导疗法代表了一种更为积极的治疗方法,从治疗管理开始就使用有效的免疫抑制药物来阻止炎症进展过程。有效的免疫抑制剂使用后,一旦疾病控制,调整为使用耐受性更好的免疫调节药物进行维持治疗。这种方法的优点在于在最短的时间内使用免疫抑制剂来实现和维持对疾病活动的控制。诱导治疗的优势是更容易更快地实现NEDA(无疾病活动证据)或最接近NEDA。诱导治疗(IT)方案包括[7]:(1)有限剂量药物:米托蒽醌、克拉德比滨、环磷酰胺;(2)持续使用药物:芬格利莫德、那他珠单抗;(3)有限时间内使用的药物:阿仑妥珠单抗、利妥昔单抗、奥克利珠单抗;(4)最终的消融剂:自体造血干细胞移植(AHSCT)。
文献指出HAMS患者关于DMT的使用,在病程早期和轻度残疾的年轻患者中效果更好[8,9]。糖皮质激素是MS复发的一线治疗方法[10,11]。糖皮质激素被认为通过发挥快速的免疫抑制作用,对MS的复发有产生作用[12]。数据表明,通过EDSS评分或Kurtzke功能系统量表的测量,使用糖皮质激素可降低复发的严重程度,并加速患者的恢复[10,11]。本例患者病程中予激素冲击、硫唑嘌呤免疫抑制、丙种球蛋白调节免疫等治疗,症状有所缓解,但仍反复复发加重,考虑患者年轻,既往未规律DMT药物治疗,2021年4月治疗上加用DMT药物西尼莫德,症状基本完全缓解,随访至2022年8月,16个月未再复发,患者一般情况可,无残疾进展。
针对本例患者临床特点,早期诊断难度较大,临床医师应提高对MS发作性症状及HAMS临床特征的认识,对早期、意外获残,频繁复发以及MRI上高活动性等显著特点,应考虑HAMS可能,因其治疗时间窗窄,更需要医师早期识别并采用合适的诱导治疗方案,以最大改善患者后期生活质量。该患者目前使用DMT西尼莫德治疗中,症状未再发,复诊一般情况可,后期继续西尼莫德治疗,仍需进一步随访评估,依据患者临床表现及影像学特点确定是否需调整治疗方案。
所有作者均声明本研究不存在利益冲突

























