
患儿,女性,1岁1个月,因"间断腹泻、便血7个月余"就诊。
患儿7个月前出现腹泻,便血,伴体重增长缓慢。体格检查:身高70 cm,体重6.0 kg,精神差,营养不良貌,毛发稀疏,皮肤干燥,皮下脂肪菲薄。口腔黏膜光滑,可见软腭裂,腹软,肝脏肋下2.0 cm,质地软,肠鸣音活跃。
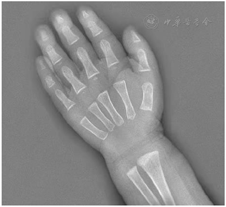
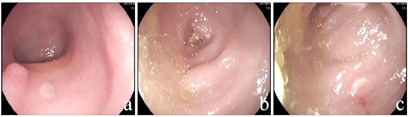
肝功能提示转氨酶升高,血常规提示中性粒细胞和血红蛋白降低。左手腕部正位X线片显示骨龄明显落后于1岁。肠镜检查发现结直肠黏膜糜烂,肠腔内可见较多油性食物残渣附着。全外显子测序显示患儿SBDS基因复合杂合突变,c.258+2T>C来自父亲,为已知致病突变,c.100A>G来自母亲,尚未报道。
给予口服胰酶、补充脂溶性维生素、对症支持等综合治疗。
患儿无腹泻、便血,身高体重增长可,现定期随访6个月,病情稳定。
儿科;消化科;血液科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
Shwachman-Diamond综合征(Shwachman-Diamond Syndrome,SDS)是一种常染色体隐性遗传病(OMIM:260400),发病率为1/7.5万,SBDS基因缺陷是主要的致病原因。SDS患者多在婴儿期起病,男女发病率比例为1.7:1[1],其临床特征主要包括胰腺外分泌功能障碍、骨骼异常和骨髓功能障碍,还可累及肠道、肝脏、神经系统、免疫系统等,并具有向骨髓增生异常综合征或急性髓系白血病等恶性转化的倾向[2]。部分患者临床特征异质性较大,常常被误诊、漏诊,延误治疗。目前国外学者有关该病的分子生物学研究已逐渐开展,国内关于该病诊治的报道不多。现报道1例SBDS基因新变异致Shwachman-Diamond综合征的病例,旨在增强临床医师对该病的认识,以早期干预,改善预后。
患儿,女性,1岁1个月,因"间断腹泻、便血7个月余"入院。患儿入院前7个月出现腹泻,每天4~8次,量中等,为黄色稀糊状便,偶有少量鲜红色血丝,无发热、腹胀、呕吐、抽搐、水肿等,给予更换深度水解配方奶粉。2个月前患儿腹泻加重,每天10余次,为黄色水样便,当地医院给予"补液、蒙脱石散及益生菌"等对症治疗,大便3~4次/d,好转出院;之后再发腹泻,过敏原检查显示牛奶蛋白过敏,给与更换氨基酸奶粉,口服益生菌、蒙脱石散等治疗,腹泻稍好转,易反复,遂来我院。近4个月体重增加0.5 kg。出生史及家族史无异常。4个月可抬头,7个月出牙,8个月可独坐,1岁1个月扶走、说"妈妈、爸爸"等简单语言,认知及智力发育正常。父母及1姐姐均体健。体格检查:身高70 cm(<P3),体重6.0 kg,血压84/58 mmHg(1 mmHg=0.133 kPa),精神差,营养不良貌,皮肤干燥,皮下脂肪菲薄,毛发稀疏。口腔黏膜光滑,可见软腭裂,腹软,肝脏肋下2.0 cm,质地软,脾脏肋下未及,肠鸣音活跃。心肺及神经系统查体阴性。
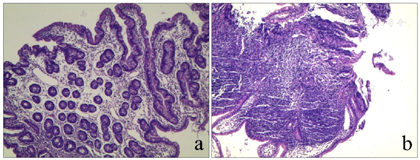
血常规:白细胞4.88×109/L,血红蛋白103 g/L,血小板219×109/L,中性粒细胞计数0.52×109/L;肝功能:谷丙转氨酶256.8 U/L,谷草转氨酶151.6 U/L。铁蛋白升高(848.7 ng/ml),血气分析、肾功能、心肌酶、电解质、血糖、三酰甘油、脂肪酶、凝血功能、体液及细胞免疫功能、铜蓝蛋白、粪常规、腹部彩超均正常,甲状腺功能:甲状腺素62.93 nmol/L,偏低,余正常。骨髓象:有核细胞减少,中性粒细胞减少,红系比例减低,血小板散在聚集可见,淋巴细胞比值增高。粪培养提示菌群失调;自身抗体谱、血尿遗传代谢筛查、巨细胞及EB病毒IgM抗体及DNA、T-SPOT、粪便艰难梭菌毒素检测、叶酸、维生素B12均阴性。左手腕部正位X线片显示骨龄明显落后于1岁(图1)。胃镜示上消化道胃镜检查未见异常;肠镜示结直肠黏膜糜烂样改变,肠腔内可见较多油性食物残渣附着(图2)。胃肠镜病理示:十二指肠黏膜绒毛低平,固有层水肿,淋巴管轻度扩张,散在淋巴细胞、浆细胞浸润。回肠末端黏膜绒毛低平,固有层脉管轻度扩张,淋巴组织增生。大肠黏膜组织慢性炎症,固有层内嗜酸性粒细胞增多,26个/HP(图3)。
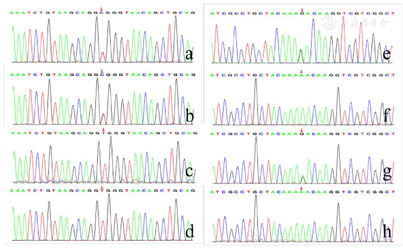
基因检测:经父母知情同意,并通过本院伦理委员会审核批准(批号:2022-K-033)后,采集先证者及其父母、姐姐外周静脉血各5 ml进行家系全外显子测序,对患儿及其父母变异位点采用Sanger测序验证确定变异来源。基因检测结果显示先证者存在SBDS基因复合杂合变异:c.258+2T>C和c.100A>G(图4),其姐姐均为野生型。Sanger测序验证结果发现,c.258+2T>C变异来源于患儿父亲,c.100A>G变异来源于患儿母亲。c.258+2T>C变异为经典剪切变异,将引起mRNA外显子拼接错误,为已经报道的热点致病突变。c.100A>G(p.Asn34Asp)为错义变异,导致SBDS基因的第34位氨基酸由天冬酰胺变为天冬氨酸,该变异在正常人群数据库未收录,经查阅Clinvar、HGMD、PubMed、中国知网、万方数据知识服务平台等数据库均未发现有该变异的报道,为首次报道。错义变异位于致病热点区(第30、31、32、33、34位氨基酸变异均为致病性变异),符合中等致病证据PM1;该变异在公共人群数据库(千人基因组数据库、EXAC数据库、ESP数据库)中频率小于0.0005,符合中等致病证据PM2;在隐性遗传病中,在反式位置上检测到致病变异,符合中等致病证据PM3;此变异之前未曾报道,但是在同一位点,导致另外一种氨基酸的变异(Asn34Ser,Asn34Ile)已有文献报道[3,4],符合中等致病证据PM5;氨基酸变异危害性评估预测软件Polyphen2、SIFT和Mutationtaster预测该变异对基因产物有害,支持致病证据PP3。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics ,ACMG)指南,c.100A>G判定为疑似致病性变异。
患儿反复腹泻,生长发育落后,营养不良,血液血红蛋白及粒细胞低,肝功能转氨酶升高,肠腔内有较多油性食物残渣附着及十二指肠、回肠末端黏膜绒毛低平,根据临床表现、血液及基因分析结果,确诊为Shwachman-Diamond综合征。
本病需要与以下疾病鉴别:(1)囊性纤维化:是由囊性纤维化跨膜传导调节因子基因突变引起的罕见的常染色体隐性遗传病,主要表现为慢性阻塞性肺疾病、胰腺功能不全及汗液钠、氯离子升高等,无骨髓衰竭表现,结合基因可明确。(2)乳糜泻:称麦胶敏感性肠病,常在6~24个月龄饮食引入麸质后出现,表现为慢性腹泻、生长迟滞、体重减轻及贫血,十二指肠黏膜萎缩,抗组织谷氨酰胺转移酶抗体阳性,HLA( Human Leukocyte Antigen ,HLA)分型具有很高的阴性预测价值,可用于排除乳糜泻。(3)食物蛋白诱导的肠病:多在出生后1岁内出现症状,临床表现为摄入可疑食物后出现呕吐、慢性腹泻,影响体质量和身高,有些患儿伴脂肪泻,肠黏膜组织学可显示隐窝增生、绒毛萎缩,回避过敏食物有效,患儿在3岁左右症状可逐渐消失。(4)先天性粒细胞减少症:是一种与多基因突变有关的罕见的遗传异质性疾病,临床上以外周血中性粒细胞绝对计数<0.5×109/L,并有严重的或反复感染为特征,还可合并皮肤、中枢神经系统、胰腺、心脏等其他系统病变,并具有向骨髓增生异常综合征或急性髓系白血病等恶性转化的风险,外周血中性粒细胞计数及全外显子测序未发现其他与患儿表型相关的致病或者可能致病突变,可排除。
患儿口服外源性胰酶,补充脂溶性维生素A、D、E、K,高热量饮食对症治疗。
现定期随访6个月,患儿无腹泻、便血,体重为8.1 kg,身高为76 cm;肝功能仍有异常:谷丙转氨酶144.1~231.5 U/L,谷草转氨酶123.7~132.1U/L;血常规血红蛋白108~113 g/L,白细胞总数与血小板正常,中性粒细胞计数0.65~1.65×109/L,维持治疗中,患儿病情稳定。
SDS是以儿童胰腺外分泌功能不全、骨髓衰竭和干骺端发育障碍为特征的常染色体隐性遗传病。约90%的病例是由SBDS基因变异引起[5]。
SBDS基因位于7q11,含5个外显子,编码250个氨基酸的蛋白,参与微管稳定、肌动蛋白聚合、核糖体生物发生过程和成熟,在骨髓细胞增殖、有丝分裂和基质微环境中发挥重要作用[6,7]。位于SBDS基因2号外显子上的无义变异Lys62Ter和移码变异C83fs变异,均可致蛋白质翻译提前终止[8]。Liu[9]等人的研究表明,SBDS通过调节端粒酶募集来维持人类端粒系统,可基于此开发SDS和其他端粒酶相关疾病的治疗靶点。目前SBDS基因最常见的3种变异形式是c.183 -184 TA> CT纯合变异、c.258+2T>C纯合变异及两个变异位点的复合杂合变异[10]。除了SBDS基因,DNAJC21基因[11,12]、EFL1基因[12]和 SRP54基因[13]变异也可导致类SDS表型,上述基因在SDS患者中所占比例均<1%。本研究中,患儿SBDS基因存在杂合变异,分别为c.258+2T>C和c.100A>G。前者为经典剪切变异,为已经报道的热点致病突变。c.100A>G变异目前未见报道,是新发现的错义变异,根据ACMG指南评级为疑似致病性变异。到目前为止,尚未观察到SDS基因型和表型之间的相关性。
SDS已报道的病例临床表现复杂多样,典型的临床症状包括胰腺外分泌功能障碍相关的吸收不良、脂肪泻和生长迟缓,单一或多种血细胞减少血液异常并骨髓增生异常综合征和急性粒细胞白血病,以及身材矮小、骨畸形、神经异常、肝功能不全、重症感染等。一项来自意大利的研究显示,粒细胞缺乏、胰腺外分泌功能不全、骨骼异常发生率分别为80%、75%和50%[14]。随着时间的推移,患者之间,甚至同一个人前后的临床表现差异很大[15]。吸收不良、脂肪泻、脂溶性维生素缺乏和生长障碍是胰腺外分泌障碍的典型临床表现,多于婴儿期起病,随年龄增长,生长迟缓更为明显。本例患儿婴儿期起病,反复腹泻,生长发育迟缓、营养不良,与胰腺外分泌功能障碍的临床表现相符。SDS患者骨骼发育异常主要表现为身材矮小,干骺端发育不良,骨龄落后,骨质疏松,全身性骨质减少,肢体畸形等[16,17]。该患儿明显身材矮小、骨龄明显落后,符合骨骼发育异常的主要表现。此外,心脏、胃肠道、肾脏等先天性畸形表现均有报道。
实验室检查显示持续或间歇性的中性粒细胞减少是最常见的血液学异常表现,80%的患者会出现贫血,网织红细胞低,24%~88%的患者会出现血小板减少[2],随病情进展可表现为全血细胞减少,并有向骨髓增生异常综合征或急性髓系白血病转化的可能性。本例患儿发病即出现两系降低,需警惕恶性转化的风险。另外,肝脏受累表现为从无症状的转氨酶升高、肝肿大、肝脏脂肪浸润到由于肝纤维化引起的不同形式的持续性慢性肝病,甚至肝硬化、肝衰竭[18]。大多数转氨酶升高患儿随年龄的增长而恢复正常,少数患儿出现转氨酶水平持续升高和肝纤维化。该患儿出现血清谷丙转氨酶升高,排除了由感染、肝毒性药物引起的肝损伤,给予保肝、补液、氨基酸奶粉营养等治疗,病情反复。结合患儿临床表现、实验室检查及基因筛查等综合分析,明确诊断为SDS。此外,除了胰腺外分泌功能障碍外,肠病在某些情况下可能导致症状,胃肠黏膜活检提示超过50%的有症状SDS患儿中,十二指肠组织学显示从绒毛钝化到绒毛萎缩及不同程度的炎性改变[19],亦有合并克罗恩病的报道[20]。本例患儿肠镜示结直肠黏膜点片状糜烂,肠黏膜病理示绒毛低平,固有层水肿,淋巴管轻度扩张,大肠黏膜组织慢性炎症,固有层内嗜酸性粒细胞增多,结合患儿有腹泻、便血,曾考虑小肠淋巴管扩张症及过敏性肠炎,给予更换氨基酸奶粉及富含中链脂肪酸的深度水解配方粉治疗效差,故临床上碰到难治性腹泻、生长发育缓慢、中性粒细胞减少、肝酶异常的病例,肠黏膜病理示绒毛低平,淋巴管轻度扩张,除外常见疾病,应考虑到SDS的可能,尽早行基因学检查明确诊断。
SDS的治疗主要包括:替代治疗、造血干细胞移植(hemopoietic stem cell transplantation,HSCT)治疗和基因治疗等。替代治疗,对于胰腺外分泌功能障碍者可给予口服胰酶及补充脂溶性维生素替代治疗。研究发现补充胰酶的需求随着年龄的增长可能逐渐减少,约一半的患者在4岁时胰腺功能可接近正常[7]。针对粒细胞减少者可适当应用粒细胞集落刺激因子,必要时预防性使用抗生素。对于发生严重的骨髓衰竭或骨髓增生异常综合征和白血病的患者,唯一的治愈性治疗是HSCT[21]。研究显示,随着年龄的增长,恶性血液系统并发症的发生风险逐渐增高,在明显的骨髓增生异常综合征或白血病发生之前,在恶性转化的早期阶段进行HSCT与更好的预后相关[21]。目前基因治疗已取得一些进展,新型翻译通读诱导药物可以促进SBDS蛋白再合成,改善骨髓造血祖细胞的髓系分化,增强中性粒细胞成熟并减少体外发育不良的粒细胞数量,在斑马鱼中显示出非常低的毒性,为今后临床基因治疗夯实基础[22]。确诊SDS的患者应定期监测血常规、凝血功能、维生素水平及骨髓细胞学。此外,还需要从口腔与牙齿、发育与神经心理评估等方面进行检测及管理患者。目前,本研究患儿正在给与替代治疗,动态监测血常规,必要时行HSCT,目前仍在定期随访过程中。
综上所述,由于遗传学、分子生物学、基因组学等的不断发展,SDS的疾病谱系逐渐扩大,对于病因不明的难治性腹泻患儿,若同时伴生长迟缓、中性粒细胞减少、肝功能异常,应考虑SDS。及时完善基因检测有助于早期明确诊断并指导及时治疗,同时为遗传咨询和优生优育提供帮助。此外,本研究发现的c.100A>G新的变异位点扩展了SBDS基因的变异谱。
所有作者均声明本研究不存在利益冲突

























