
患儿,男性,3岁8个月,因"肥胖2年,睡眠增多3个月,多饮多尿及高血压1个月"就诊。
患儿以快速进展的肥胖起病,逐渐出现日间睡眠增多,猝倒发作,夜间睡眠时中枢性低通气,多饮多尿,高血压、低热及性格改变。查体示体型均匀性肥胖,清醒安静时呼吸尚平稳,睡眠时呼吸浅,双下肢远端皮温低。
患儿有快速进展的肥胖,下丘脑功能障碍,中枢性低通气,自主神经功能障碍等表现,睡眠增多伴猝倒发作,下丘脑分泌素<110 pg/ml,多次小睡实验阳性,PHOX2B基因片段分析及全外显子基因测序阴性。故诊断为ROHHAD合并发作性睡病。
低盐低脂饮食,限制热卡,积极运动,调整饮食及改变生活习惯以减轻体重;睡眠时应用无创正压通气辅助呼吸,监测呼吸、经皮氧饱和度情况,调整无创呼吸机参数以避免严重二氧化碳潴留;口服降压药等对症处理;因年龄偏小,无治疗发作性睡病药物的适应症,暂未予药物治疗。
患儿于我院出院时日间精神较前有所好转,睡眠期无创正压通气辅助呼吸下经皮氧饱和度维持在90%左右,二氧化碳潴留好转。出院后3个月因经皮氧饱和度不能维持,于当地医院行气管插管连接呼吸机辅助呼吸支持治疗。
儿科;神经内科;内分泌及遗传代谢科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
快速发作性肥胖伴通气不足、下丘脑功能障碍、自主神经失调(Rapid-onset Obesity with Hypoventilation, Hypothalamic Dysfunction, Autonomic Dysregulation,ROHHAD)综合征是一种罕见疾病,至今全世界报告了大约100例。ROHHAD综合征是一类多器官疾病,该疾病的特点是早期和快速发作的肥胖,并伴有通气不足、自主神经调节失调和内分泌异常[1],可并存神经源性肿瘤。本文报道1例以体重迅速增加为首发表现且累及多器官系统的典型ROHHAD综合征病例,提高临床医生对本病的认识,以早期诊断,减少并发症的发生。
患儿,男性,3岁8个月,因"肥胖2年,睡眠增多3个月,多饮多尿及高血压1个月"于2022年3月就诊于北京儿童医院。
病史:入院前2年(即1岁8个月),患儿无明显诱因出现体重明显大于同龄儿(具体体重不详),入院前1年(即2岁8个月),患儿体重19 kg,入院前4个月(即3岁4个月),患儿体重28 kg,之后发现体重增长迅速,每月约增加2 kg。入院前3个月,患儿无明显诱因出现睡眠增多,具体表现为日间及夜间睡眠时间均增多,无明显打鼾及张口呼吸,无睡眠中不自主运动及行为异常。入院前2个月,患儿出现行走时遇到障碍物易摔倒。入院前1个月,患儿睡眠较前明显增多,且快速入睡,似"倒头即睡",唤醒较困难,每日基本于床上活动。患儿出现多饮、多尿,饮水量1500~2000 ml/d,饮水后反复打嗝,尿量具体不详,夜尿增加至5~6次/夜,就诊于当地医院,住院第4天,患儿睡眠12小时后无法唤醒,伴小便失禁,伴口唇及甲床紫绀,呼吸浅弱,经皮氧饱和度下降,予鼻导管吸氧后经皮氧饱和度升至90%左右,可疑抽搐1次,表现为双眼上翻、双手握拳并抽动,予咪达唑仑静推后缓解,但仍有呼吸浅慢,经皮氧饱和度不能维持,遂转至PICU,予气管插管连接呼吸机辅助通气,经皮氧饱和度可维持在95%以上,并予甘露醇降颅压、头孢哌酮舒巴坦抗感染、阿昔洛韦抗病毒、甲泼尼龙冲击等治疗,期间间断发热4 d,体温最高38.4℃,应用退热药物后体温可降至正常;监测血压98-124 mmHg/56-67 mmHg(1 mmHg=0.133 kPa),予卡托普利口服后血压降至100/80 mmHg左右。呼吸机辅助通气3 d后予拔管,监测经皮氧饱和度85%~90%,予低流量吸氧后血氧维持在95%左右。入院前9 d,患儿情绪激动或害怕后出现发作性点头、发呆、双眼斜视,数秒即过,爱发脾气。入院前7 d,患儿上述症状不缓解,伴眼神发呆、发飘,突然双下肢无力致经常性向前跌倒,意识清楚,呼之可应,不愿爬楼梯及行走,就诊于我院门诊。自发病以来,患儿精神反应可,不愿与人交流,但可与父母正常交流,体力较差,懒动。既往史:无特殊。出生史:G2P2,因其母为"瘢痕子宫"足月剖宫产,出生体重3.6 kg,生后无窒息缺氧史,新生儿期体健。生长发育史:智力及体力发育同正常同龄儿,3个月开始抬头,4个月会翻身,6个月会坐,8个月会爬,11个月会站,12个月会走,12个月会说话。家族史:否认家族性遗传病史。
内科查体:血压126/77 mmHg,体重32.0 kg(大于同性别同年龄儿童第97百分位),身高104 cm(位于同性别同年龄儿童第75百分位),BMI 29.6 kg/m2(大于同性别同年龄儿童第97百分位),体型均匀性肥胖,上下部量及肢体比例正常,腹部可见皮肤紫纹,全身皮肤未见黑棘皮,皮肤颜色及发色正常,未见牛奶咖啡斑及色素脱失斑。清醒安静时呼吸尚平稳,胸廓活动度尚可,节律规整,无发绀,睡眠时呼吸浅快。心脏及腹部查体大致正常。双下肢远端皮温低,双侧足背动脉搏动可。
神经系统查体:头围52 cm,无畸形。神志清楚,表情淡漠,问话不答,但与其母亲交流尚可,步态正常。双眼睑无下垂,双侧瞳孔等大等圆,对光反射存在。构音清,饮水无呛咳,伸舌无偏斜,颅神经查体大致正常。四肢肌力V级,肌张力正常,腱反射正常引出,病理征阴性,深浅感觉正常,脑膜刺激征阴性,共济查体正常。
辅助检查:
(1)PHOX2B基因片段分析:受检者PHOX2B基因(GCN)n重复次数为16和20次,为正常型。
(2)全外显子组(WES)检测CNV:未检出可以解释临床表型的单核苷酸变异或微小缺失/重复变异,未发现与受检者临床症状相关的明确致病性的大片段缺失/重复变异。

(1)多导睡眠监测:患儿清醒时血氧平均87%,经皮二氧化碳分压平均91.7 mmHg。入睡后非快速眼动睡眠期平均血氧78%,经皮二氧化碳分压96.5 mmHg,快速眼动睡眠期平均血氧75%,经皮二氧化碳分压94.6 mmHg。予持续低流量氧气吸入0.5L/分,平均血氧97%,经皮二氧化碳分压107 mmHg。患儿夜间体温波动在37.5~37.7℃,呼吸浅快,平均60~80次/min,吸氧前后无明显变化。无张口呼吸,无鼾声,以中枢型低通气事件为主。
(2)多次小睡实验:平均睡眠潜伏期1.8 min,可见4次快速眼动睡眠,快速眼动睡眠潜伏时间1.4分钟。结果:阳性。
(3)脑脊液下丘脑分泌素:62.54 pg/ml(正常值>110 pg/ml)。
(1)脑脊液常规、生化、墨汁染色、抗酸染色、肺炎支原体抗体、一般细菌培养、TORCH-IgM:未见异常。
(2)脑脊液细胞学、血及脑脊液抗神经元抗体检测(NMDA+GAD+VGKC)、血及脑脊液抗神经元抗体检测(Ri+Hu+Yo):未见异常。
(3)头颅核磁平扫:左侧侧脑室稍增宽,双侧侧脑室旁白质轻度T2 FLAIR高信号,余未见异常。
(1)动态血压监测:全天平均血压114/74 mmHg,白天平均血压115/75 mmHg,夜间平均血压121/72 mmHg,全天平均收缩压及舒张压偏高,白天平均收缩压及舒张压偏高,夜间平均收缩压及舒张压偏高。血压昼夜节律减弱。
(2)促肾上腺皮质激素-皮质醇节律:均正常范围内,节律紊乱。正常节律呈昼夜周期性变化,晨起含量最高,下午逐渐降低,至夜间将至最低(表1)。
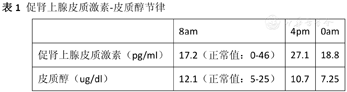
促肾上腺皮质激素-皮质醇节律
促肾上腺皮质激素-皮质醇节律
| 8am | 4pm | 0am | |
|---|---|---|---|
| 促肾上腺皮质激素(pg/ml) | 17.2(正常值:0-46) | 27.1 | 18.8 |
| 皮质醇(ug/dl) | 12.1(正常值:5-25) | 10.7 | 7.25 |
肾素-血管紧张素-醛固酮系统:肾素偏高,血管紧张素II及醛固酮正常(表2)。
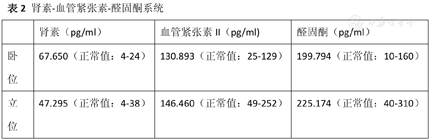
肾素-血管紧张素-醛固酮系统
肾素-血管紧张素-醛固酮系统
| 肾素(pg/ml) | 血管紧张素II(pg/ml) | 醛固酮(pg/ml) | |
|---|---|---|---|
| 卧位 | 67.650(正常值:4-24) | 130.893(正常值:25-129) | 199.794(正常值:10-160) |
| 立位 | 47.295(正常值:4-38) | 146.460(正常值:49-252) | 225.174(正常值:40-310) |
(4)心率减速力分析:心率减速力>2.5ms,<4.5ms,提示迷走神经调节能力下降。心率加速力>-2.5ms,>-4.5ms,提示交感神经调节能力下降。
(5)动态心电图:窦性心律,窦性心律不齐,部分窦性心动过速,平均心率128次/分,可见ST抬高。
(6)心脏彩超、血管超声、肾上腺超声:未见异常。
(1)甲功五项:血清游离T4 8.13 pmol/L,稍低,余正常。
(2)甲状腺抗体三项、空腹胰岛素及C-肽、糖化血红蛋白、生长激素、生长因子、尿儿茶酚胺、血氨、乳酸、维生素B12、血清叶酸、同型半胱氨酸、铜蓝蛋白:未见异常。
(3)血尿代谢筛查:血Pip偏高伴尿乙基丙二酸略高。
(4)甲状腺超声:未见异常。
(5)垂体核磁:垂体上缘凹陷,垂体稍变扁。
(1)脑电图:背景活动略慢;双侧顶、枕、后颞导联散发慢波;监测家长指认事件(点头、发笑、表情异样、偏头),同期脑电图未见明确相关异常放电。
(2)血常规:正常,无红细胞及血红蛋白升高。
(3)肿瘤标志物:甲胎蛋白、癌胚抗原、人绒毛膜促性腺激素、神经元特异性烯醇化酶未见异常。
(4)肺CT及气道重建:两肺背侧胸膜下散在间实质病变,可见小结节影,胸膜略厚,大气道重建未见异常。
(5)腹部超声:胰腺脂肪浸润,余未见异常。
ROHHAD合并发作性睡病:根据患儿为3岁8个月余学龄前男童,隐匿起病,出生时正常,出生后1岁6个月内无明显相关症状,1岁8个月起体重明显较同龄儿重(具体体重不详),2岁8个月体重19 kg,3岁4月体重28 kg,之后发现体重增长迅速,每月约增加2 kg,3岁5个月出现睡眠增多,睡眠时呼吸浅快,存在明显二氧化碳潴留及低氧,无打鼾、张口呼吸,肺CT及气道重建未见异常,心脏彩超未见异常,结合多导睡眠监测,提示睡眠相关中枢性低通气。患儿存在肥胖、多饮多尿、中枢性发热、促肾上腺皮质激素升高及促肾上腺皮质激素-皮质醇节律紊乱等下丘脑功能障碍,存在高血压、双下肢末梢皮温低,饮水后连续打嗝,心率减速力分析提示迷走神经、交感神经调节能力下降等自主神经功能障碍。另外,患儿有性格暴躁、不愿与人交流等性格改变。结合其PHOX2B基因片段分析及CNV正常,故诊断快速发作性肥胖伴通气不足、下丘脑功能障碍、自主神经失调(ROHHAD)综合征。本病50%~70%合并神经源性肿瘤,本患儿查神经元特异性烯醇化酶、尿儿茶酚胺正常,肺CT、腹部超声、肾上腺超声、睾丸超声等未见明显异常,目前未发现肿瘤征象。根据患儿日间睡眠明显增多,且入睡迅速,似"倒头即睡",唤醒较困难,可疑猝倒发作(情绪激动或害怕后出现发作性点头、发呆,或突然双下肢无力致跌倒,意识清楚,呼之可应),易说梦话,同时伴有肥胖、呼吸障碍,不愿与人交流等表现,多次小睡实验提示平均睡眠潜伏期1.8 min,可见4次快眼动睡眠,快眼动睡眠潜伏时间1.4 min,结果阳性,结合其脑脊液下丘脑分泌素62.54 pg/ml,<110 pg /ml,故诊断发作性睡病。综上,患儿诊断为ROHHAD综合征合并发作性睡病。
鉴别诊断:
本病是以中枢呼吸系统及自主神经系统控制障碍为特征的一种罕见病,是PHOX2B基因突变引起的常染色体显性遗传疾病。该疾病发病年龄通常在新生儿期,但也可能在儿童期或成年期被诊断。患者主要表现为呼吸暂停、低氧血症和高碳酸血症,且在睡眠期间最为严重,尤其是非快速眼动睡眠。PHOX2B基因突变导致呼吸中枢化学感受器的原发性缺陷,导致对二氧化碳敏感性降低,从而出现一系列临床症状。PHOX2B在在正常等位基因上会有一段20个丙氨酸的重复片段,大多数患者丙氨酸重复次数将增加为24~33次,而约10%具有非丙氨酸重复扩展突变,包括错义、无义或移码突变。
本病为自身免疫反应介导的脑炎,部分患儿有前驱感染史,临床主要表现为精神症状、认知损害、意识水平下降、抽搐、语言障碍、运动障碍、睡眠障碍、自主神经功能障碍或中枢性低通气等。早期以精神症状或认知损害、抽搐常见,而后期以意识水平下降、运动障碍、自主神经功能障碍常见。此类疾病最常见为抗N-甲基-D-天冬氨酸受体脑炎。本病脑脊液白细胞升高或寡克隆区带阳性,脑电图示局灶性或弥散性慢波、电活动紊乱、癫痫波或极端δ刷,头颅核磁正常或非特异性异常。本患儿以睡眠障碍、自主神经功能障碍、可疑抽搐为主要表现,需考虑本病可能,血、脑脊液特异性抗体阳性具有鉴别诊断意义。
低盐低脂饮食,限制热卡,积极运动,调整饮食及改变生活习惯以减轻体重;睡眠时予无创正压通气辅助呼吸,先予BPAP-S(Bilevel Positive Airway Pressure-Spontaneouns,双水平气道正压通气自主触发)模式,入睡后呼吸频率浅快,约60~70次/min,事件以中枢型低通气为主伴血氧下降,平均血氧92%,经皮二氧化碳分压80 mmHg,同时给予氧疗,未见明显改善,后改予BPAP-S/T(Bilevel Positive Airway Pressure-Spontaneous /Timed,双水平气道正压通气自主触发时间控制)模式,同时给予1L/分氧疗,平均血氧95%,经皮二氧化碳分压67 mmHg,R期仍间断出现血氧下降,经整夜压力滴定,给予治疗压力为BPAP S/T模式,13/5 cmH2O(1 cmH2O=0.098 kPa),呼吸频率25次/min,吸气时间0.8 s;监测呼吸、经皮氧饱和度情况,于我院睡眠中心多次调整无创呼吸机参数,以使二氧化碳分压逐渐降低;口服降压药等对症处理;因年龄偏小,无治疗发作性睡病药物的适应症,且患儿经予呼吸支持后日间睡眠时间较前缩短,故暂未予药物治疗。
患儿于我院出院时精神反应较前好转,语言及活动较前增多,日间睡眠时间较前缩短,睡眠期无创正压通气辅助呼吸下经皮氧饱和度维持在90%左右,动脉血气分析示二氧化碳分压自入院时82 mmHg降至52 mmHg,二氧化碳潴留较前好转。患儿出院后2个月,症状再次加重,出现睡眠增多,伴口唇、甲床发绀;出院后3个月,因经皮氧饱和度不能维持,于当地医院行气管插管连接呼吸机辅助呼吸支持治疗。
ROHHAD综合征一种罕见疾病,由Fishman等[2]于1965年首次描述,并于2007年由Ize-Ludlow等[3]重新命名为ROHHAD,其首字母缩写表明症状发展的顺序。ROHHAD综合征一种复杂疾病,需多学科协作以明确诊断与治疗方向,涉及到神经内科、耳鼻喉科、内分泌科、呼吸科、营养科、心脏内科、肿瘤科及神经外科等科室。
本病的发病机制尚不明确,目前有三种主要的发病机制假说,包括遗传理论、表观遗传理论和自身免疫理论[1]。遗传学研究调查了神经元发育、下丘脑和自主神经功能障碍通路的候选基因,包括HTR1a、OTP、PACAP、HCRT、HCRTR1和HCRTR2,但没有发现任何显著的遗传变异[4]。Patwari等[5]报道了同卵双胞胎其中一个患有ROHHAD综合征,而另一个正常,表明同卵双胞胎表观基因组变异导致双胞胎表型不一致的可能性。有文献报道应用大剂量免疫抑制剂,如环磷酰胺、利妥昔单抗、免疫球蛋白等进行免疫抑制治疗有良好治疗效果,印证了ROHHAD综合征的自身免疫发病机制[6]。此外,ROHHAD综合征患儿的尸检结果显示以淋巴细胞浸润为特点的脑炎,同样支持免疫介导的发病机制[7]。
ROHHAD综合征的发病年龄在2至4岁之间,伴有食欲过盛和体重迅速增加[8],在这些症状出现前,患儿的生长发育及一般情况均正常。Julie Harvengt等[9]对43例ROHHAD患者的临床症状及预后进行了系统回顾,研究表明快速肥胖作为首发症状,随后有83%的患者在开始肥胖后的前5年内被诊断为中枢性通气不足。通气不足有可能危及生命,所有患者都接受了夜间无创通气治疗,避免夜间低氧血症及肥胖加重。本例患儿睡眠中曾出现1次突发口唇及甲床紫绀,入院后行多导睡眠监测示患儿低氧血症睡眠期较清醒期加重,且在快速眼动睡眠阶段更为严重,平均血氧饱和度为75%,经皮二氧化碳分压为94.6mmHg,通气不足是ROHHAD综合征中最危及生命的症状,因此呼吸系统异常可以增加对ROHHAD综合征的早期识别[10]。在首发症状后数月至数年内会出现下丘脑功能障碍及自主神经失调。下丘脑功能障碍包括高催乳素血症、甲状腺功能减退、生长激素缺乏症、中枢性性早熟、高钠血症、低钠血症、烦渴或尿崩症。自主神经失调包括眼科方面异常,如视力模糊、瞳孔对光的反应改变、斜视、上睑下垂,胃肠动力受损,如腹泻、便秘,体温失调,疼痛感知改变,出汗过多,手脚冰冷等[11]。此外,约40%的ROHHAD综合征可伴有神经源性肿瘤,如星形胶质细胞瘤、节细胞神经母细胞瘤等[12],发病率相对较高,因此怀疑患有ROHHAD综合征的患者也需要进行筛查。本患儿筛查神经元特异性烯醇化酶、尿儿茶酚胺正常,胸部CT、腹部超声、肾上腺超声、睾丸超声等未见明显异常,目前未发现肿瘤征象,需要定期复查肿瘤标志物及影像学。
由于没有特殊的辅助检查结果支持ROHHAD综合征的诊断,目前诊断基于以下临床表现:(1)从儿童早期开始的快速肥胖和睡眠期间的通气不足;(2)下丘脑功能障碍和自主神经失调的体征和症状;(3)排除引起类似特征的其他疾病,例如先天性中枢性低通气综合征。快速发作的肥胖和最常见的内分泌疾病,如性早熟和甲状腺功能减退症通常是早期可识别的迹象[1]。本病与先天性中枢性低通气综合征均表现为内分泌系统异常及自主神经失调,但后者存在PHOX2B基因突变,可通过完善基因检测作为鉴别诊断的要点。本患儿以快速进展的肥胖、睡眠时中枢性低通气、多饮多尿等下丘脑功能障碍及高血压、低热、末梢皮温减低等自主神经功能障碍为临床症状,PHOX2B基因片段分析及CNV正常,符合ROHHAD综合征诊断标准。此外,患儿日间睡眠增多明显,且入睡迅速,可疑猝倒发作,结合其多次小睡实验阳性及脑脊液下丘脑分泌素结果,考虑ROHHAD综合征合并发作性睡病。
本病死亡率高达50%-60%,主要因通气不足、心肺衰竭和心肺骤停导致患儿死亡[13],早期识别和干预可以最大限度地降低死亡率。
综上,本文报道了1例罕见且复杂的ROHHAD综合征合并发作性睡病的病例,本患儿的多种临床症状及体征对该疾病起到早期识别的作用。随着社会经济发展和生活水平的提高,儿童肥胖比例显著升高,当2岁左右儿童存在快速肥胖、通气不足、内分泌疾病、下丘脑功能障碍等表现,在诊断时需警惕ROHHAD综合征可能。对该疾病的早期诊断,会改善预后,减少并发症的发生。
所有作者均声明本研究不存在利益冲突

























