
患者,女性,7岁。因卵巢肿瘤破裂导致的腹痛行开腹左附件切除术,术中未见卵巢外转移病灶。术后病理提示幼年性颗粒细胞瘤,予以博来霉素、依托泊苷及卡铂化疗6程。3个月后患者出现盆腹腔多发转移并再次行手术切除,术后病理仍考虑为幼年型颗粒细胞瘤盆腔腹腔广泛种植,二代测序提示EWSR1 exon12-CREM exon6融合。再次予以化疗9程,但3个月后病情再次复发。病情中激素六项、AMH及血清CA 125等肿标一直均正常
复发后患者无明显临床症状。
进行术后病理诊断,包括形态学、免疫组化和分子特征。
患者入院后行腹腔镜探查+开腹肿瘤细胞减灭术,肿瘤广泛种植于盆腹腔,术后病理显示上皮样细胞被纤维间质分隔呈分叶状生长,肿瘤细胞异型性明显,免疫组化提示SOX10,S-100,INI-1及α-inhibin阳性。最终诊断为EWSR1 exon12-CREM exon6融合的卵巢上样恶性周围神经鞘瘤。患儿恢复后按照恶性周围神经鞘瘤予以基于大剂量多柔比星联合异环磷酰胺的化疗方案开始化疗。
规律随访3个月,患者的一般情况尚可,但复查增强CT提示1程化疗后病情稳定,患者目前仍在接受周期为4周的同方案的化疗中。
妇产科;骨与软组织科;肿瘤科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
恶性周围神经鞘瘤(Malignant peripheral nerve sheath tumor,MPNST)是一种罕见的恶性肿瘤,发病率为百万分之一人年,约占软组织肉瘤的5%~10%[1,2,3]。MPNST恶性程度高,大约40%的患者在5年内发生肿瘤转移。MPNST患者的5年总生存率(overall survival,OS)为51%~62.1%,5年无进展生存率(progress-free survival, PFS)为37%~52.9%[4,5,6,7,8]。手术治疗是MPNST的主要治疗策略,完全切净肿瘤能最大程度改善患者的预后,而术后辅助治疗如放化疗的作用和预后影响因素尚未有一致的结论[5,6,7]。
上皮样恶性周围神经鞘瘤(Epithelioid malignant peripheral nerve sheath tumor,EMPNST)是MPNST中的一种独特的病理亚型,其特点是上皮样肿瘤细胞占主要成分,呈现出多小叶生长模式并伴有黏液样和/或纤维间质,仅有部分病例可见肿瘤细胞呈纺锤状形或梭形[9]。尽管EMPNST仅占MPNST的2.5%~6.2%[5,10],但其预后相对更差[11]。此外,ESWR1-CREM基因融合的恶性上皮样肿瘤好发于间皮内衬的腹膜腔,并倾向于出现多发盆腹腔膜膜转移的生物学行为特征[12]。卵巢性索间质肿瘤(Ovarian sex-cord stromal tumors, OSCT)占所有原发性卵巢恶性肿瘤的近7%,其中颗粒细胞瘤是最常见的亚型[13]。幼年型颗粒细胞肿瘤通常表达抑制素[14],有趣的是,这是一种在ESWR1-CREM基因融合的上皮样肿瘤中也表达的免疫组化标记物[12]。此外,EMPNST误诊为OSCT可能会导致较差的生存结局,因为这两种疾病的治疗方案上存在显著差异。
发生于女性生殖系统的MPNST极为罕见,目前英文文献报道仅有约20例[15],而卵巢原发的MPNST至今未见报道。我们报道1例7岁女童的OEMPNST合并EWSR1-CREM基因融合的病例,以研究这种极其特殊的病例的临床特征,并复习文献讨论其治疗方案。
患者,女性,7岁,因"卵巢性索间质肿瘤二次术后化疗后,发现复发1个月"入院。2020年8月17日行开腹左附件切除术,术中发现肿瘤术前已破裂,大小约8.5 cm×7 cm×5.5 cm,无远处转移。术后病理:符合幼年型颗粒细胞瘤,免疫组化:α-Inhibin(+),CD99(+),Ki-67(约20%+)。术后予以博来霉素+依托泊苷+卡铂静脉化疗6程(末次为2021年1月19日)。
2021年5月7日出现第一次复发,2021年5月19日行开腹盆腹腔多发肿物切除+腹膜后肿物切除+大网膜切除术,术中见病灶主要位于肝肾隐窝、侧腹膜、髂窝、结肠系膜、小肠壁、盆腔壁等,部分经后腹膜延伸至腹膜后区域,包绕髂血管;左卵管残端、大网膜可见多发肿物。手术医院的病理仍考虑为幼年型颗粒细胞瘤盆腔腹腔广泛种植。NGS:EWSR1 exon12-CREM exon6融合,NF2基因缺失。但另外两家医院会诊考虑诊断:符合EWSR1-CREM基因融合的上皮样恶性外周神经鞘瘤;加做免疫组化提示:SOX-10,S-100及H3k27me3阳性。尽管如此,患儿仍被按照颗粒细胞瘤复发予以化疗9程(长春新碱+异环磷酰胺+依托泊苷2程、白蛋白紫杉醇+异环磷酰胺+顺铂7程,末次化疗2021年12月18日)。2022年4月6日复查超声提示盆腹腔多发病灶,进一步行盆腹腔MRI及PET/CT评估均考虑病情复发,病灶分布于盆腔腹膜、残余大网膜及肠道表面。病程中未见异常肿瘤标记物、性激素及AMH水平。余既往史、家族史、个人史无特殊。
入院前复查肿瘤标记物及性激素六项、AMH均在参考值范围内。2022年4月10日MRI:左侧腰大肌前方3枚结节,直径约8 mm,转移可能。腹部散在结节影,肿瘤腹膜转移可能,肠系膜根部散在小淋巴结。4月26日PET/CT:胃窦下方、左肾前方、脾下方、右上腹肠系膜上、左腰大肌前方、左中腹大网膜上多发转移灶,较大者约1.2 cm×0.8 cm(图1)。其余血常规、肝肾功能、尿常规、凝血、血型、输血8项及心电图等术前检查、检验基本正常。
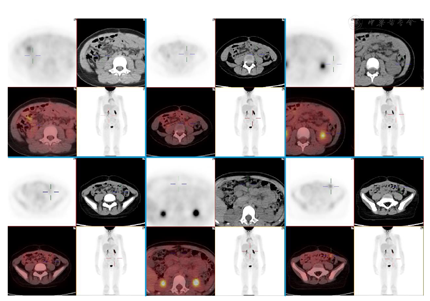
结合患者的病史、体征、辅助检查,起初考虑卵巢性索间质肿瘤,幼年型颗粒细胞瘤可能性大,需与卵巢上皮性良性肿瘤、交界性或恶性上皮性卵巢肿瘤鉴别;结合分子特征及免疫组化,最终诊断为卵巢上皮样恶性周围神经鞘瘤(OEMPNST)。
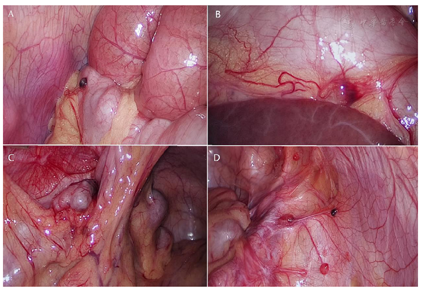
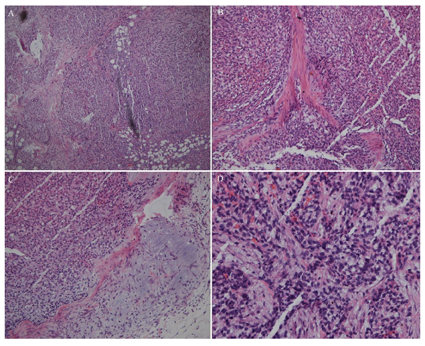
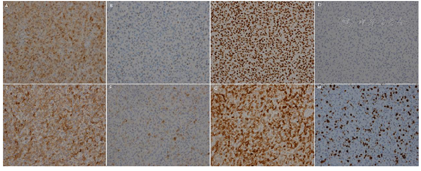
2022年5月5日在全麻下行腹腔镜探查+开腹再次肿瘤细胞减灭术(盆腹腔转移瘤切除+大网膜切除+横膈肿物切除术,探查见:子宫直肠窝、小肠系膜、乙状结肠及系膜、升结肠、回盲部及腹壁腹膜、后腹膜、侧壁腹膜均可见散在结节。残余部分网膜上可见散在肿瘤结节,较大者2 cm。降结肠侧沟可见散在小结节,肠壁表面肿瘤结节,大者约2 cm;横膈双侧可见淡红色肿瘤结节,较大者约1.5 cm。肾周包膜散在结节(图2)。未探及肿大淋巴结。予以逐次切除前述腹水少量。手术彻底切净,横膈及腹膜表面少许粗糙面予氩气烧灼。手术切除绝大部分肉眼可见病灶,术后病理:卵巢上皮样恶性周围神经鞘瘤,广泛累及(前腹壁;膀胱表面;子宫后壁;左侧骨盆漏斗韧带;乙状结肠系膜;膈肌肿物;小网膜表面;脾区肿物;右肾包膜;横结肠系膜表面;大网膜;乙状结肠壁;升结肠系膜;右侧腹膜;小肠系膜);免疫组化结果:AE1/AE3(+),Calretinin(散在+),GFAP(+),EMA(-),CD56(散在+),Ki-67(index 50%),Melan-A(-),SOX10(散在+),S-100(+),α-inhibin(+),WT-1(浆+),INI-1(+)(图3,图4)。因考虑最终诊断为OEMPNST而非OSCT,按照MPNST的诊治指南,与患儿家长商讨化疗方案,建议行大剂量多柔比星联合异环磷酰胺的方案辅助化疗。因患儿家长顾虑,手术后约2个月才开始按照恶性周围神经鞘瘤予以基于大剂量多柔比星联合异环磷酰胺的化疗方案开始第一次化疗。
患者术后恢复顺利,化疗前复查增强CT提示病情进展,1程化疗后复查影像学疗效评估为稳定。规律随访3个月,患儿一般情况尚可,化疗后骨髓抑制较为严重,仍在接受周期为4周的同方案的化疗中。
我们报道1例7岁儿童的EWSR1-CREM基因融合的OEMPNST,最初被误诊为OSCT。通过生物学特征、形态学、免疫组化染色、分子特征等最终诊断,提示全面的评估对正确诊断卵巢肿瘤至关重要。
MPNST是指发生于周围神经或神经外软组织的恶性肿瘤,伴有神经鞘分化。一半以上的MPNST为散发性疾病,既往研究中有11.7%~51%的病例与神经纤维瘤病-1 (Neurofibromatosis-1,NF-1)相关,其余10%为放疗诱发[4,5,6,16]。MPNST好发于40~50岁人群,无性别差异。肿瘤主要起源于四肢、躯干、头部和颈部,而原发生于腹部或内脏的MPNST非常罕见,仅占所有MPNST的1%[5],这可能是MPNST在腹部或内脏肿瘤的鉴别诊断中经常被忽视的原因之一。
S-100、SOX-10和胶质纤维酸性蛋白(Glial fibrillary acid protein, GFAP)是诊断MPNST常用的免疫组化标记物,但既往研究显示,MPNST的S-100、SOX-10和GFAP阳性表达率分别约占40%、30%~67%和30%[17,18]。在另一纳入63例EMPNST的研究中,所有的EMPNST均表达S-100,其中87%为强阳性和弥漫阳性[9]。此外,几乎所有的(91.7%~100%)EMPNST均表达H3K27me3(H3K27 trimethylation)[10,19],显微镜下EMPNST表现为多小叶生长模式,其中呈小叶状和巢状的肿瘤细胞被粘液样和/或纤维间质所分隔包绕[9],这表明它在形态学上与MPNST不有所同。我们的病例与这些结果一致,具有典型的形态学表现且肿瘤组织表达H3K27me3、SOX-10和S-100,强烈支持EMPNST的诊断。
AFT1、CREB1 CREM (cAMP-responsive element modulator, cAMP反应元件调制器)构成CREB转录因子家族。编码FET家族RNA结合蛋白和CREB转录因子家族如EWSR1和FUS的基因融合已被证明在恶性软组织肿瘤和间皮瘤的肿瘤发生机制中发挥重要作用[20,21]。最近,Argani等[12]发现EWSR1-CREM基因融合定义了一系列独特的恶性上皮样肿瘤,这些肿瘤好发于具有间皮衬覆的盆腹腔中。在这个研究中,13例患者中有11例的肿瘤起源于间皮衬覆的盆腹腔或出现腹腔内转移,绝大多数病灶累及大网膜、结肠系膜或直肠阴道隔。EWSR1-CREM基因融合的肿瘤具有转移至盆腔的恶性生物学潜能,7例患者诊断时即出现转移或后续进展为肿瘤转移[12]。此外,具有EWSR1-CREM基因融合的肿瘤可表达抑制素和细胞角蛋白(AE1/AE3),易误诊为OSCT[12]。由于OSCT的好发年龄和免疫组化发现肿瘤组织中α-抑制素和calretinin的高表达,这里患儿在初次治疗和出现第一次复发时也被误诊为OSCT。然而遵照OSCT的标准治疗方案对该例患者的治疗效果非常差,短期内即出现复发,表现为广泛的盆腹腔种植转移。免疫组化中SOX-10、S-100、INI-1及H3K27me3的表达排除了OSCT的可能性,α-抑制素的表达可能与该肿瘤具有EWSR1-CREM融合基因有关。
MPNST是一种侵袭性强、预后差的肿瘤,约2/3的患者会出现疾病进展或复发,近50%的患者会在确诊后5年内死亡,该病在成人和青少年中的生存结局无显著差异[5,8]。预后因素包括肿瘤大小、肿瘤分级、横纹肌肉瘤组间研究(Intergroup Rhabdomyosarcoma Study,IRS)组别和NF-1表达与否[4,5,7]。然而,NF-1的表达状态对生存结局的影响仍存在争议[5,7,16]。
目前,手术是MPNST患者的主要治疗策略,但术后肿瘤残留病灶大小与预后显著相关,更高的残留肿瘤负荷提示显著更差的预后[4,5,6,7]。在一个儿童MPNST的队列中,IRS分组Ⅰ组、Ⅱ组、Ⅲ组和Ⅳ组的5年OS和5年PFS分别为82%和61%、62%和37%、32%和27%、26%和21%[7]。研究显示,在MPNST患者中,13.8%至74%的患者接受了化疗,38%至75.7%的患者接受放疗[4,5,7]。然而,化疗和放疗在改善临床预后方面的效果存在争议[8,22]。曾有报道显示辅助放疗可降低术后仍有微小残留肿瘤病灶患者的局部复发率,而化疗可提高手术完全切除肿瘤的可能性[7,23]。Carli等[7]报道了11例儿童MPNST患者在新辅助化疗后实现了手术完全切除肿瘤,尽管他们在最初的评估中被认为肿瘤病灶无法完全切除。此外,在MPNST患者中,以阿霉素为基础的方案是首选化疗方案,大剂量阿霉素联合异环磷酰胺方案的缓解率最高[3,6,7]。
原发于女性生殖道的MPNST非常罕见,目前英文文献仅报道21例[15,24]。其中,宫颈是最常见的受累部位,见于2/3(14/21)的患者,其次为外阴(7/21)。初次诊断时仅有3例患者合并转移,另有7例患者在2年内出现复发。手术是主要的治疗手段。在明确报道了临床结局的女性生殖道原发MPNST的18例患者中,4例患者死亡,5年OS率为61.4%,这与发生于其他更常见部位的MPNST患者相当[4,5]。与起源于女性生殖道其他部位的MPNST不同,我们的患者发病更早,初次就诊时患儿年龄仅为5岁。据我们所知,这是第一例起源于卵巢的MPNST。经历2次复发后最终确诊,由于起源于卵巢、病灶肉眼外观类似于幼年颗粒细胞肿瘤,且存在α-抑制素的表达导致了误诊的发生。这可能为妇科肿瘤增加了一个新的概念。
综上,EWSR1-CREM基因融合的OEMPNST容易因表达α-抑制素而误诊为OSCT,综合肿瘤的生物学特性、形态学、免疫组化和分子特征在内的评估对于准确诊断及鉴别OSCT和OEMPNST至关重要。OMEPNST的治疗原则可以参考MPNST,但囿于样本量,有必要进一步研究其临床特征、治疗反应和生存结局。尽管OEMPNST极其罕见,妇科肿瘤医生仍需考虑该诊断的可能性,特别是当考虑为OSCT且经规范治疗但效果仍欠佳时。
所有作者均声明本研究不存在利益冲突

























