
患儿,女性,2岁4月龄,因"发现转氨酶升高2个月余"就诊至我院,病初外院抗体1:320,双链DNA阳性;余相关辅助检查大致正常,就诊我院给予保肝治疗后好转,但逐渐出现血小板减低,为进一步诊疗收入院系统评估。患儿3月龄发现中性粒细胞减低,外院基因检测提示KRAS c.C64A(p.Q22K)新发变异,给予升白细胞药物治疗后恢复正常。
入院后查体示生长发育正常,双腋、双侧腹股沟多发肿大淋巴结,质韧,活动可,心肺腹查体正常。
结合患儿病史、症状体征,考虑RAS相关自身免疫性白细胞增殖性疾病(RAS-associated autoimmune leukoproliferative disease,RALD)可能性大,行淋巴结穿刺活检提示淋巴组织反应性增生。行KRAS基因验证患儿血液有突变,头发和指甲无变异,明确诊断为RALD。
加用激素、西罗莫司治疗,病情稳定后激素逐渐减量,定期随访。
随访1年,患儿查体未及明显肿大的浅表淋巴结,血小板、转氨酶均正常范围。
儿科 风湿免疫科 血液科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
RAS基因相关自身免疫淋巴增殖性疾病(Ras-associated autoimmune leukoproliferative disorder, RALD)是由KRAS、NRAS基因突变引起的一组罕见的原发性免疫缺陷病[1]。该病临床表现多样,其常见的临床特征包括肝脾大、淋巴组织增生、自身免疫性血细胞减少、B细胞或单核细胞增多症、高丙球蛋白血症等,该病易继发恶性肿瘤[2]。本文总结了1例KRAS基因突变致RALD患者的临床资料及基因检测结果,并对其诊疗进行探讨,旨在提高临床医生对RALD的认识。
患儿,女性,2岁4月龄,因"发现转氨酶升高2个月余"就诊。患儿病初因常规体检发现转氨酶升高:ALT 177U/L,AST 95 U/L。外院完善血常规:WBC 9.35×109/L,N 2.93×109/L,HGB 129 g/L,PLT 281×109/L;CRP、ESR、补体、免疫球蛋白等均大致正常;肝炎病毒系列、EB病毒、细小病毒B-19等均阴性;抗核抗体1:320;自身免疫性肝炎抗体谱、血管炎抗体、抗磷脂抗体谱等自身抗体均阴性;腹部超声:肝脾大小径线正常范围,肝门及脾门区多发淋巴结增大,余未见明显异常;先天性代谢病血尿筛未见异常。为进一步诊疗就诊我院,加用保肝药物治疗4个月左右监测血小板逐渐减低,最低至48×109/L。为系统评估收入院治疗。
患儿系第一胎第一产儿,母亲孕期体健。患儿生长、语言、运动及智力发育等均大致正常。患儿生后3月龄发现中性粒细胞减低(N 0.31×109/L,血WBC、PLT、Hb均正常范围),外院行基因检测提示KRAS c.C64A(p.Q22K)新发变异,白血病相关基因及核型未见异常,给予升白细胞药物治疗后恢复至正常范围。
体格检查:体重16.3 kg(>P97),身高97.5 cm(>P97),头围52 cm(>P97)。精神反应好。后背皮肤散在陈旧出血点。双腋下触及肿大淋巴结,最大者2.0 cm×1.0 cm,质韧,活动可;双侧腹股沟可触及肿大淋巴结,最大2.0 cm×1.0 cm,质韧,活动可。心肺腹无殊,肝脾肋下未及。关节活动好,四肢肌力、肌张力正常。
入院后完善相关检查:血常规:WBC 6.98×109/L,N 2.82×109/L,Hb 127g/L,PLT 54×109/L;尿便常规、肾功、CRP、ESR、维生素B12水平均正常;肝功:AST 98U/L,ALT 172 U/L;细小病毒B19-IgM阳性,余病毒系列均(-);外周血细胞形态分析:淋巴细胞增生;免疫球蛋白:IgG 31.11 g/L,IgG亚类均明显升高;抗核抗体1:640,双链DNA、Coombs试验均阳性,自身免疫性肝炎抗体谱:SLA、AMA-M2均阳性;TB细胞亚群大致正常;复查补体C3、C4均减低;IL-10 35.5 pg/ml;双阴性T细胞检测(CD4CD8 double-negative T cell,DNT):3.82%(合96个/ul,升高);I型干扰素水平测定:18.069(升高);铜蓝蛋白、甲胎蛋白均正常范围。
淋巴结超声:双侧腋窝、双侧颈部多个低回声淋巴结,皮质增厚,皮髓质分界可见。PET/CT:双侧上颈深部、颈后部、左锁骨上、双肺及纵膈、双侧腋窝、肝门、脾门、腹主动脉周围多发代谢增高淋巴结。腹部超声、肝静脉及下腔静脉超声均未见明显异常。
骨穿:增生尚活跃,粒:红=6.69:1,粒系中、晚幼粒细胞、红系晚幼红细胞比例减低,形态大致正常;粒细胞占46.0%,比例升高,可见少许幼稚淋巴细胞占4.5%;单核细胞比例增高,形态正常;全片共计巨核细胞5个,血小板数量减少。
患儿病程中逐渐出现慢性淋巴结增生、转氨酶升高、血小板减低、多种自身抗体阳性、补体减低、外周血DNT细胞及多克隆性IgG水平增高,结合基因检测,考虑RALD可能性大。
鉴别诊断方面:(1)部分RALD患者可有类似系统性红斑狼疮(systemic lupus erythematosus,SLE)的表现, KRAS基因突变也可导致单基因儿童SLE,应注意鉴别[3,4]。依据2019年EULAR和ACR分类标准[5],该患儿抗核抗体>1:80,存在双链DNA、Coombs试验均阳性,补体C3、C4均减低,合并血液系统受累。符合SLE的诊断标准,SLEDAI评分为5分(PLT减低,低补体血症及双链DNA阳性,为轻度活动)。但该患儿起病年龄较小,肝酶明显升高,DNT细胞水平增高,且存在脾脏、淋巴结增大等淋巴系统增殖性疾病,与儿童系统性红斑狼疮相比,该患儿症状相对不典型,因此需进一步检查明确病因。(2)RALD既往被认为是自身免疫性淋巴细胞增生综合征(autoimmune lymphoproliferative syndrome, ALPS)-IV型,但RALD不存在FAS、FASL或CASP10的生殖细胞系或体细胞突变;此外,RAS基因的生殖细胞突变,除引起肿瘤外,还可引起Noonan综合征等疾病,该病可表现为生长发育迟缓、特殊面容、先天性心脏病等;该患儿无生长发育落后,无特殊面容、先天性心脏病等异常,可进一步行基因验证,明确是否为体细胞突变。(3)该病亦应与幼年型粒单核细胞白血病(juvenile myelomonocytic leukemia,JMML)相鉴别,JMML是婴儿和儿童罕见的侵袭性骨髓增殖性/骨髓增生异常性疾病,表现为外周血、骨髓和内脏中异常粒单核细胞浸润增加;多数JMML患者亦存在RAS/MAPK信号通路基因的体细胞突变和/或种系突变,可伴有自身免疫性疾病,临床表型与RALD重叠;目前该患儿外周血及骨穿均未见异常粒单核细胞浸润增加,故不支持JMML。RALD易继发恶性肿瘤,因此诊断过程中亦需排查淋巴瘤可能。综上我院建议患儿完善淋巴结活检,同时行KRAS基因突变体细胞验证明确基因变异情况。
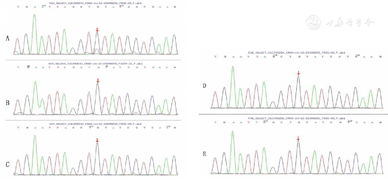
患儿淋巴结活检病理符合淋巴组织反应性增生。对KRAS基因进行验证(图1),患儿血液存在KRAS基因突变,但患儿头发、指甲及患儿父母外周血细胞相同基因位点均无突变,结合患儿临床特征、突变预测、突变位点正常人频率及突变类型等,提示患儿存在KRAS基因体细胞突变,可明确诊断为RALD。
患儿明确诊断后在保肝治疗基础上加用激素(1 mg/kg)及西罗莫司口服,病情稳定后激素逐渐减量,定期监测患儿淋巴结、肝脾大小,检测血常规、转氨酶、自身抗体、补体、免疫功能等的变化。
患儿规律使用激素及西罗莫司后10 d左右血小板升至正常范围至今,转氨酶逐渐降至正常范围,后激素逐渐减量。目前随诊1年余,激素减量至5 mg qd维持,继续应用西罗莫司治疗。患儿生长发育均正常范围,查体未及明显肿大的浅表淋巴结;血小板稳定在300×109/L左右,转氨酶正常范围;超声检查提示肝脾大小正常,肝区淋巴结消失,双腋窝及脾区淋巴结均较前明显缩小。
RALD的发病机制主要为KRAS和NRAS基因增功能突变导致RAF-MEK-ERK和PI3K-AKT-mTOR信号通路活化增强,进而影响淋巴细胞的增殖、分化、存活及凋亡[6,7]。既往文献认为RALD可合并I型干扰素通路的激活[8]。该患儿I型干扰素水平测定高于正常范围,与既往文献相符。RALD临床表现多变,可表现为反复感染、脾大、淋巴组织增生、单核细胞增多症、高丙种球蛋白血症和自身免疫性血细胞减少,也可表现为SLE等自身免疫性疾病[1,8,9,10]。既往报道3例以早发性狼疮样综合征起病的RALD患儿,其均为NRAS基因突变,并对RALD的诊治总结相关经验:对于疑诊RALD的患者,首先检测血液样本的NRAS或KRAS突变;若存在NRAS或KRAS突变,则检测毛囊、唾液、指甲和口腔黏膜细胞等部位是否存在突变,以确认突变为体细胞[10]。
2021年Neven等总结了RALD相关的基因异常,其中KRAS基因突变位点包括p.G12A、p.G12D、p.G12S、p.G13A、p.G13C、p.G13D等[11]。该患儿临床表型与RALD临床表型一致,经验证发现存在KRAS基因的血液突变,但指甲、毛发无突变,结合其临床表现、相关实验室检查、相关治疗反应等,可明确诊断为RALD。
RALD需依据病情严重程度给予个体化治疗,若患者表现为单纯淋巴结肿大、肝肿大、脾肿大者建议密切随访;若合并严重自身免疫性疾病症状首选激素治疗,可联合吗替麦考酚酸酯或西罗莫司控制病情;多数患者治疗后病情稳定[8,12,13],对于难治性自身免疫性血细胞减少症,激素及其他免疫抑制剂治疗无效者,可应用利妥昔单抗治疗[14]。对于无明确恶性病变证据的RALD患者,建议避免行造血干细胞移植[2]。该患儿临床表现为SLE,且存在脾大等淋巴细胞增生性表现,经激素联合西罗莫司治疗后患儿症状逐渐改善,考虑激素联合西罗莫司治疗有效,尚无应用利妥昔单抗、造血干细胞移植等治疗的指征。
生殖细胞系RAS突变与特定的发育障碍相关,包括Noonan综合征、Costello综合征等,而体细胞RAS突变见可于多发性骨髓瘤、JMML等侵袭性肿瘤及RALD等疾病中[1,3]。JMML患者可表现为脾大、发热、血小板减少、单核细胞增多以及浸润皮肤和重要器官的过多粒-单核细胞[2]。KRAS和NRAS基因突变导致JMML的发生风险增加。目前尚不能确定RALD是一种非恶性疾病,还是一种癌前状态,还是一种儿童期克隆性惰性肿瘤[2]。既往文献报道1例KRAS基因突变患儿病初表现为RALD,15年后进展为侵袭性白血病[15]。因此建议对RALD患者进行密切监测。本研究中患儿随访1年,各项指标趋于好转,但其远期预后仍待进一步随访。
综上,对于以自身免疫性血细胞减少、高丙种球蛋白血症、伴慢性淋巴结增生的早发型SLE样综合征患者应注意完善基因检测,警惕RALD的可能;激素、西罗莫司对于合并自身免疫性疾病症状者有效,治疗过程中应长期随访,警惕JMML等恶性疾病的发生。
所有作者均声明本研究不存在利益冲突

























