
3例女孩,初诊年龄初诊年龄7.1岁~8.8岁,均因身材矮小,8岁前出现乳房发育先后就诊,有矮小家族史。
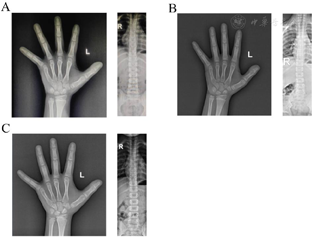
3例患儿均表现为身材矮小(-2.81SD~-2.24SD),例2伴有特殊面容(高额头、低鼻梁、中面部发育不良)、短颈、大拇指短,例1、例3无面容及骨骼异常。
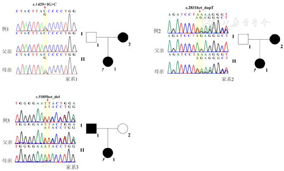
生长激素激发试验峰值分别为8.6 ng/ml、9.3 ng/ml、16.8 ng/ml。促性腺激素释放激素激发试验示促黄体生成素峰值分别为7.8 IU/L、10.4 IU/L、6.6 IU/L,子宫0.9~1.5 ml,卵巢1.1~1.6 ml,骨龄提前1.2~2.9岁,确诊中枢性性早熟;全外显子测序和外显子捕获实验提示3例患儿均携带ACAN基因杂合致病性变异:例1携带ACAN剪接变异:c.1429+1G>C,p.Val465Glyfs*13(母源);例2、3均携带ACAN移码变异:c.2831dupT,p.Glu945Argfs*484(母源),c.5185delA,p.lle1729Tyrfs*2(父源);所有变异均为新发现的变异。
3例患儿均予重组人生长激素(rhGH)单用或联合促性腺激素释放激素类似物(GnRHa)治疗。
例1予rhGH治疗12个月,身高增加8.5 cm,ΔHtSDS:0.74SD;例2予rhGH治疗2个月后予联合GnRHa治疗14个月,身高增加11.5 cm,ΔHtSDS:0.62SD;例3予rhGH联合GnRHa治疗21个月,身高增加10.2 cm,ΔHtSDS:0.44SD。
儿科;内分泌与遗传代谢科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
ACAN基因(OMIM :165800)位于15q26染色体上,编码聚集蛋白聚糖(aggrecan)的产生,是矮小症的致病基因之一。Aggrecan是一种硫酸软骨素蛋白聚糖,是软骨细胞外基质的关键成分,在骨骼发育过程中起着关键作用。ACAN基因变异的临床特征主要包括身材矮小、面部异常和骨骼发育不良,ACAN基因变异合并性早熟较为少见,目前仅有2例报道[1,2]。现报道3例以矮小、乳房发育提前为主要临床表现的ACAN基因变异患儿临床资料,并评估重组人生长激素(recombined human growth hormone, rhGH)单用或联合促性腺激素释放激素类似物(Gonadotropin-releasing hormone analogue,GnRHa)治疗疗效,旨在提高对本病的认识,做到早诊断、早治疗。
例1,女孩,7岁7个月,以"发现身材矮小6年余,双侧乳房发育1 d"为主诉就诊。患儿系G1P1,足月剖宫产,母孕期无特殊用药史,出生体重(BW):3.0 kg,出生身长(BL):50 cm。查体:身高108.8 cm(-3.33 SD),体重20.0 kg,无特殊面容,双侧乳房Tanner分期II期(B2),女童外阴,阴毛Tanner分期I期(PH1),阴蒂外观正常,无阴唇融合,脊柱、四肢未见异常。父亲身高173 cm,母亲身高143 cm(-3.26 SD)。
例2,女孩,7岁1个月,以"发现身材矮小6年余,双侧乳房发育半年"为主诉就诊。患儿系G1P1,足月剖宫产,母孕期无特殊用药史,BW:3.7 kg,BL:50 cm。查体:身高112.0 cm(-2.24 SD),体重22.5 kg。特殊面容(高额头、低鼻梁、中面部发育不良),短颈,大拇指短,双侧乳房B2,女童外阴,阴毛PH1,阴蒂外观正常,无阴唇融合。脊柱稍侧弯,无其他骨骼异常。父亲身高166 cm,母亲身高144 cm(-3.25 SD),外祖父身高155 cm(-3.11 SD)。
例3,女孩,8岁10个月,以"发现身材矮小8年余,双侧乳房发育1年"为主诉就诊。患儿系G1P1,足月剖宫产,母孕期无特殊用药史,BW:3.4 kg,BL:51 cm。查体:身高117.2 cm(-2.81 SD),体重23 kg,无特殊面容,双侧乳房B2,女童外阴,阴毛PH1,阴蒂外观正常,无阴唇融合,脊柱稍侧弯,无其他骨骼异常。父亲身高153 cm(-3.46 SD),母亲身高146 cm(-2.86 SD)。
所有患儿血尿常规、肝肾功能、甲状腺和肾上腺皮质功能均正常。3例患儿均予生长激素(growth hormone,GH)激发试验,其中2例患儿GH部分缺乏(GH峰值<10 ng/mL);促性腺激素释放激素(Gonadotropin releasing hormone,GnRH)兴奋试验提示3例黄体生成素(Luteinizing hormone,LH)峰值>5 IU/L,染色体正常。上腹部和妇科彩超未见异常,骨龄提前,脊柱X线片提示3例患儿脊柱无明显异常,垂体MRI未见异常。3例患儿主要临床资料见表1。

3例患儿临床资料
3例患儿临床资料
| 项目 | 病例1 | 病例2 | 病例3 | 正常值 |
|---|---|---|---|---|
| 性别 | 女 | 女 | 女 | |
| 年龄 | 7岁7月 | 7岁1月 | 8岁10月 | |
| 身高 | 108.8 cm(-3.33SD) | 112.0 cm(-2.24SD) | 117.2 cm(-2.81SD) | |
| GH峰值 | 8.59 | 16.80 | 9.27 | >10.00 ng/ml |
| IGF-1 | 217 | 327.0 | 212.0 | 49.0~297.0ng/ml |
| IGFBP3 | 4.34 | 6.32 | 4.83 | 0.70~10.00 mg/L |
| LH基础值 | 0.10 | 0.10 | 0.13 | 0~2.30 IU/L |
| FSH基础值 | 1.15 | 1.72 | 1.71 | 0.11~1.60 IU/L |
| LH峰值 | 7.78 | 10.40 | 6.59 | <5 IU/L |
| FSH峰值 | 13.20 | 16.80 | 7.82 | <5 IU/L |
| E2 | <5 | <5 | <5 | 0~5 pg/ml |
| T | <2.5 | <2.5 | <2.5 | 0~2.5 ng/dl |
| 骨龄 | 8岁10月 | 10岁 | 10岁2月 | |
| 妇科彩超 | 子宫(0.91 ml) | 子宫(1.45 ml) | 子宫(1.29 ml) | |
| 卵巢(1.46 ml/1.14 ml) | 卵巢(1.25 ml/1.53 ml) | 卵巢(1.43 ml/1.37 ml) | ||
| 头颅MRI | 正常 | 正常 | 正常 | |
| 基因检测 | c.1429+1G>C | c.2831dupT | c.5185delA | |
| (ACAN) | p.Val465Glyfs*13 | p.Glu945Argfs*484 | p.lle1729Tyrfs*24 |
GH:生长激素;IGF-1:胰岛素样生长因子1;IGF-BP3:胰岛素样生长因子结合蛋白-3;LH:促黄体生成素;FSH:卵泡刺激素;E2:雌二醇;T:睾酮;BA:骨龄
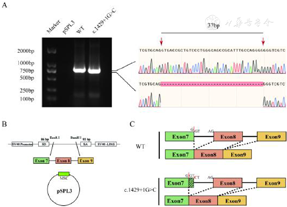
经知情同意,予患儿行全外显子测序,父母行Sanger测序验证。结果示3例患儿均携带ACAN基因变异:例1携带ACAN剪接变异:c.1429+1G>C(杂合),变异来源于母亲(杂合)。例2携带ACAN移码变异:c.2831dupT,p.Glu945Argfs*484(杂合),变异来源于母亲(杂合)。例3携带ACAN移码变异:c.5185delA,p.lle1729Tyrfs*24 (杂合),变异来源于父亲(杂合)。为了验证ACAN c.1429+1G>C变异对剪接的影响,构建了mini gene模型,转染HEK293T细胞进行表达。以空pSPL3质粒载体作为阴性对照,WT作为阳性对照。琼脂糖凝胶电泳结果显示,WT与c.1429+1G>C的cDNA片段大小有轻微差异。c.1429+1G>C的特征是外显子7存在37 bp缺失。因此,c.1429+1G>C变异导致ACAN蛋白(Val465Glyfs*13)移码。3种变异均为新发现的变异,结合美国医学遗传学与基因组学学会指南分析为致病性变异(例1:PVS1+PM2+PP1+PP4;例2:PVS1+PM2+PP1+PP4;例3:PVS+PM2+PP4)。
3例患儿均表现为严重身材矮小,8岁前出现双侧乳房发育,GnRH激发试验提示下丘脑-垂体-性腺轴功能启动,骨龄提前,但患儿没有出现线性生长加速,考虑合并其他因素,故行全外显子检测,结果提示3例患儿均存在ACAN基因杂合致病性变异。结合临床表现及基因检测结果,"矮小症(ACAN基因变异)、中枢性性早熟(central precocious puberty,CPP)"诊断明确。
3例患儿甲状腺激素正常,垂体MRI正常,可排除甲状腺功能减退及中枢神经系统异常引起的矮小和CPP。还需与其他矮小相关综合征及继发性疾病引起的性早熟相鉴别:
1.Noonan综合征[3]主要表现为身材矮小、特殊面容,但该疾病常表现为青春发育延迟,还常伴多系统发育异常,如先天性心脏病、男性隐睾等,基因变异以PTPN11、SOS2常见,与此病例不符。
2.Prader-Willi综合征[4]在儿童期亦表现为矮小,伴有GH缺乏等内分泌代谢异常,但常合并过度摄食、肥胖、认知和行为障碍等表现,是由父源性15q11-13区域基因功能缺陷引起多系统异常,可鉴别。
3.McCune-Albright综合征[5]亦可表现为性早熟,但还常具有皮肤咖啡斑、多发性骨纤维发育不良等特点,其性发育过程与CPP不同,促性腺激素水平低下,Gs基因缺陷所致,可排除。
3例患儿均予rhGH单用/联合GnRHa治疗。例1予rhGH治疗12个月后,身高117.3 cm(-2.59SD),ΔHtSDS:0.74SD;例2予rhGH治疗2个月,身高增长1.9 cm,由于骨龄增长过快,予联合GnRHa治疗14个月,身高121cm(-1.62SD),ΔHtSDS:0.62SD;例3予rhGH联合GnRHa治疗21个月,身高127.4cm (-2.37SD),ΔHtSDS:0.44SD。2例患儿接受治疗期间定期规律随访,IGF-1较前升高,性激素水平较前下降,子宫、卵巢体积较前减小,骨龄生长速度下降,未见药物不良事件。
随着基因检测技术的发展,越来越多身材矮小的遗传原因逐渐被认识,尤其是在特发性矮小(Idiopathic Short Stature,ISS)患儿中。ACAN基因变异是导致矮小症发生的原因之一,在初步诊断为ISS病例中,约占1.4%~6%[6]。ACAN基因编码的aggrecan核心蛋白包含3个球状结构域(N端G1和G2结构域,C端G3结构域)、1个球状间结构域(IGD)、1个糖胺聚糖附着区(GAD),其中GAD包括硫酸角蛋白结构域(KS)和硫酸软骨素结构域(CS)。ACAN基因在纵向生长调控中起关键作用。由ACAN基因编码的aggrecan是细胞外基质的重要成分,它通过G1与透明质酸、软骨连接蛋白相互作用,通过G3结构域上的C型凝集素重复序列(CLD)与细胞外蛋白聚糖结合,形成aggrecan聚集体。CS和KS的广泛硫化和与透明质酸的聚集产生了大量的固定负电荷,使aggrecan高度水化,由此产生的膨胀压力赋予软骨承载性能[7,8],因此,该基因在骨和软骨的形态形成中起着关键作用。ACAN基因变异使aggrecan聚集体发生变化,软骨细胞外基质结构改变,从而影响骨与软骨的生长发育,导致了矮小的发生。
杂合ACAN基因变异导致的临床表型广泛,常表现为身材矮小,高额头,低鼻梁,中面部发育不良,短颈,短指,桶状胸及早发性骨关节炎等。骨龄提前在ACAN基因杂合变异患者中较为常见,少数患儿可表现为骨龄延迟或正常骨龄[9]。ACAN基因变异临床表现常不典型,除早期生长停止,常无其他特征性临床表现,难以早期发现,故易被误认为单纯的ISS。文中3例患儿均在8岁前出现乳房发育,结合GnRH激发试验和妇科超声检查确诊CPP。值得注意的是,ACAN基因变异合并性早熟少见,包括本研究,目前仅有5例报道。该机制尚不明确,虽然近年来有研究表示aggrecan聚集体也在周围神经元网中形成,通过蛋白的相互作用调节突触的可塑性[10],但aggrecan是否影响下丘脑-垂体-性腺轴的稳定,目前尚不清楚;同时,尚不能摄入排除具有雌激素活性的环境内分泌干扰物的可能性[11]。ACAN基因变异合并性早熟可能为偶然现象,需要更多的病例进一步研究。
治疗上,对于ACAN基因合并CPP的病例,早期诊断和治疗能够在骨骺愈合前提供治疗空间和时间。包括文中3例在内,共报道5例[1,2]女孩予rhGH单用/联合GnRHa治疗,治疗前身高SD值为-2.67(-3.33~-2.25),平均治疗时间12个月,治疗后身高SD为-2.09(-2.59~-1.62),平均增加0.57±0.17SD,表明早期rhGH联合GnRHa治疗能改善ACAN基因变异合并性早熟患儿身高,但5例患儿尚未到达终身高,因此仍需要今后大样本量的随机对照研究结果验证。
综上,文中报道3例以矮小、乳房发育提前为首诊的ACAN基因新变异患儿,丰富了基因型。临床上身材矮小、骨龄提前、伴或不伴有特殊面容应考虑该病可能,基因检测可明确诊断。若出现乳房发育/其他性征应行相关检查以明确诊断。rhGH单用/联合GnRHa短期治疗可有效改善ACAN基因变异合并CPP患儿身高。
所有作者均声明本研究不存在利益冲突

























