
患者,女性,48岁,因"双下肢水肿2年,再发1个月"于2018年8月入住本院肾内科。
慢性病程,临床以蛋白尿、血尿、水肿、高血压等慢性肾炎综合征伴肾功能不全为表现。肾穿刺病理活检,光镜下表现为肾小球系膜区均质样无细胞中-重度增生,基底膜弥漫增厚为特点。电镜下可见电子致密的、散在或束状聚集的、直径43~65 nm、规律排列有横纹的纤维状物,在增宽的系膜区、系膜旁区及内皮下积聚。
经免疫组织化学证实为Ⅲ型胶原纤维,临床诊断胶原Ⅲ型肾病明确。
临床在给予血管紧张素转化酶抑制剂基础上,加用甲泼尼龙并联合吗替麦考酚酯。
尿蛋白未获缓解,肾功能逐渐进展。
肾内科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
胶原Ⅲ型肾病,又称为Ⅲ型胶原肾小球病或胶原纤维性肾小球病(CG),是一种由于Ⅲ型胶原纤维在肾小球异常沉积而致病的肾小球疾病,于1979年由日本学者Arakawa等[1]首次报道。现对国内外有关文献进行复习,并结合我院确诊患者1例,以期提高临床肾内科医师对该病的认识。
患者,女性,48岁,因"双下肢水肿2年,再发1个月"于2018年8月入住海南省人民医院肾内科。患者2年前无明显诱因出现反复双下肢水肿,呈对称凹陷性,晨轻暮重,伴有解泡沫尿,无解肉眼血尿,无脱发、口腔溃疡,无皮疹、关节痛,无畏寒、发热,就诊于当地医院,测量血压升高(具体不详),检查尿蛋白3+,考虑"慢性肾炎",予药物治疗(具体不详),浮肿可消退。但患者治疗不规律,上述症状反复发作,多次复查尿蛋白3+。1年前复查尿蛋白3+、潜血-,血肌酐133.6 μmol/L,血白蛋白35 g/L,自行口服药物治疗(具体不详),后未再复查。1个月前患者双下肢浮肿再发,性质同前,至我院门诊查尿蛋白3+、潜血+-,血尿素11.41 mmol/L、肌酐110.0 μmol/L,血白蛋白38.9 g/L,血红蛋白82 g/L。门诊以"慢性肾炎综合征"收入我科进一步诊治。病程中无多饮、多食、多尿、消瘦,无畏寒、双手细颤,无发热、关节疼痛、皮疹、光过敏,无胸闷、胸痛、呼吸困难、咳粉红色泡沫痰。既往有"高脂血症",未治疗。否认糖尿病、冠心病等慢性病病史。否认肝炎、结核等传染病病史。否认重大外伤、手术及输血史。否认药物及食物过敏史。个人史、月经史、婚育史及家族史均无特殊。
入院体格检查:体温36.7℃,脉搏77次/min,呼吸17次/min,血压156/98 mmHg(1 mmHg=0.133 kPa)。贫血貌。颜面部未见浮肿,睑结膜稍苍白。心肺腹未及阳性体征。双下肢对称性轻度凹陷性水肿。
血常规:白细胞6.71×109/L,红细胞3.1×1012/L,血红蛋白82 g/L,血小板299×109/L。尿常规:蛋白3+、潜血1+,尿镜检红细胞4个/HP。24 h尿蛋白定量3.58 g/24h。生化检查:尿素12.07 mmol/L,尿酸533 μmol/L,肌酐115 μmol/L,白蛋白32.1 g/L,甘油三酯1.33 mmol/L,胆固醇6.64 mmol/L。
自身免疫相关检查(抗核抗体、抗双链DNA抗体、ANCA抗体、抗GBM抗体、补体二项、免疫球蛋白五项、抗心磷脂抗体)、肿瘤相关检查(多种肿瘤标志物、尿本周氏蛋白、血尿轻链)、感染相关检查(乙肝、丙肝、梅毒、艾滋病)、代谢相关检查(甲亢七项、血脂、空腹血糖、餐后2 h血糖、HbA1c)均未见异常。
泌尿系彩超:左肾103 mm×50 mm,右肾97 mm×42 mm,双肾实质回声稍增强。双肾血供丰富。腹部彩超、心脏彩超未见异常。
肾脏病理:光镜,患者于2018年8月27日进行肾穿刺。肾活检光镜下检查(图1)显示:3条皮髓交界肾组织,共25个肾小球。肾小球呈分叶状,肾小球系膜区中至重度增宽,系膜区可见均质样物质沉积,基底膜增厚。毛细血管袢开放欠佳,囊壁无增厚。系膜区可见少量嗜复红物沉积。肾小管上皮细胞颗粒变性,蛋白管型形成,肾小管小灶状萎缩(20%),基底膜增厚。肾间质小灶状淋巴细胞及单核细胞浸润,伴纤维化(20%)。小动脉未见明显异常。刚果红染色阴性。免疫荧光:8个肾小球,IgA、IgM、C3、C4、C1q、IgG、FRA、κ、λ、HBsAg、HBcAg均为阴性。
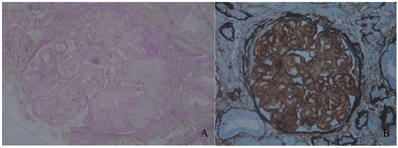
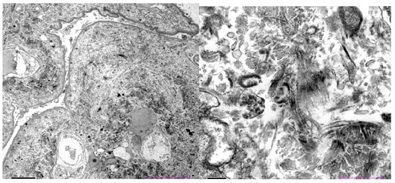
电镜检查(图2):镜下检测到1个肾小球。毛细血管内皮细胞明显空泡变性,个别管腔内可见红细胞,无明显内皮细胞增生,毛细血管襻部分受压。肾小囊壁层增厚、分层,壁层细胞空泡变性,无明显增生。基底膜:节段性增厚,厚度300~700 nm,节段性皱缩。脏层上皮细胞:上皮细胞肿胀,空泡变性。足突融合情况观察不清。系膜区:系膜细胞和基质增生。未见电子致密物沉积。系膜区及基底膜内侧可见大量规律排列有横纹的Ⅲ型胶原纤维。肾小管-间质:部分肾小管萎缩。肾间质淋巴、单个核细胞浸润伴胶原纤维增生。肾间质血管:个别毛细血管管腔内见红细胞。免疫组化(图3):肾小球Ⅲ型胶原(+),Ⅰ型胶原(-)。符合Ⅲ型胶原肾病。
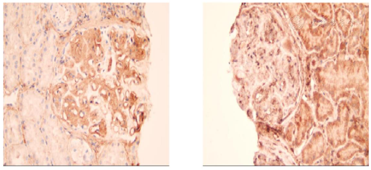
根据免疫组化结果,诊断:胶原Ⅲ肾病。
2018年8月诊断明确后加用甲泼尼龙20 mg每日1次,联合贝那普利10 mg每日1次。
门诊监测血肌酐93~96 μmol/L、尿常规:蛋白2~3+、潜血+-。2019年1月患者自行停用甲泼尼龙。2019年1月—12月未规律就诊、不规律检查提示血肌酐由102逐渐升高至126 μmol/L,尿蛋白2~3+。2020年1月—11月,未再返院就诊,未正规治疗。2020年12月来院复查肌酐221,蛋白3+,予加用甲泼尼龙20 mg每日1次联合吗替麦考酚酯0.5 g每日2次;后再门诊规律就诊,复查肾功能、尿蛋白无好转,遂于2021年5月开始逐渐减量至停用甲泼尼龙及吗替麦考酚酯。2022年6月复查肌酐289,蛋白3+。
胶原Ⅲ型肾小球病,由日本学者于1979年根据病理特点首次以"特发性系膜变性肾小球病"报道。后因电镜发现该病在肾小球系膜区、基底膜存在特殊胶原纤维沉积,改称为胶原纤维性肾小球病、胶原纤维沉积性肾病。1988年经免疫组化发现肾小球内沉积的为Ⅲ型胶原纤维,故1991年有学者建议改名为胶原Ⅲ型肾小球病。1995年被世界卫生组织正式收录进肾小球疾病的分类中。目前全球见诸文献报道的病例累计约100例[2,3,4],大部分报道来自亚洲人群,主要来自日本,其次为我国,美国、意大利、法国、印度等地有散发病例报道,现今仍属于一种临床罕见疾病。
Ⅲ型胶原蛋白可见于大多数组织的间质和血管,但通常不出现在肾小球中。在胶原Ⅲ型肾小球病的患者中,发现Ⅲ型胶原在肾小球的系膜和内皮下异常沉积。胶原Ⅲ型肾小球病目前发病机制不明,它曾被认为是指甲髌骨综合征的一种类型。指甲髌骨综合征是一种因转录因子突变引起的常染色体显性遗传疾病,其肾脏表现与胶原Ⅲ型肾小球病相似。但区别是,指甲髌骨综合征临床多表现为多系统受累,除了肾脏,还可累及指甲、膝盖骨、皮肤、眼睛等。但胶原Ⅲ型肾小球病的患者多无肾外系统的受累。并且,两种疾病的肾脏病理中,Ⅲ型胶原蛋白的沉积部位也不相同。电镜显示,胶原Ⅲ型肾小球病的Ⅲ型胶原主要在系膜区、内皮下沉积。而在指甲髌骨综合征中,Ⅲ型胶原主要在肾小球基底膜内沉积[4]。故目前认为这是两种不同的疾病。
目前关于胶原Ⅲ型肾小球病是一种原发性肾小球疾病还是全身性疾病的肾脏受累仍无定论,具体发病疾病尚不明确。目前大多认为该病的发生与胶原代谢异常有关,与患者体内Ⅲ型胶原合成和降解失衡,导致过多的Ⅲ型胶原前体释放入血,随血循环滤过到肾小球内所致。也有学者认为各种原因引起的肾小球损伤,及因此导致的细胞因子的增生、刺激,使得肾小球系膜细胞生成Ⅲ型胶原可能是该病发病的原因。在某些慢性肾小球疾病,比如系膜增生性肾小球肾炎中观察到Ⅲ型胶原增生,但大多表现轻微,且多是局灶、节段的增生,不同于胶原Ⅲ型肾小球病的弥漫增生的表现。同时有研究观察显示,由于补体调控H因子的缺陷,使得补体途径调控的异常导致肾小球内皮损伤,使得Ⅲ型胶原进入内皮下并异常沉积,可能参与了胶原Ⅲ型肾小球病的发病[7,8]。
胶原Ⅲ型肾小球病临床表现无特异性,主要表现为蛋白尿、水肿、高血压,后期逐渐出现肾功能损伤,可逐渐进展至终末期肾病[2,5]。疾病的确诊主要依靠肾组织病理检查。
该病肾脏病理的主要表现为系膜区明显增宽,以系膜基质增生为主,系膜细胞一般无增生,重度增生时可表现为结节状硬化,形似糖尿病肾病K-W结节形态;毛细血管基底膜弥漫增厚,双轨征形成;内皮下和系膜区大量弱PAS阳性物质沉积,常使毛细血管腔受压闭塞。免疫荧光常阴性,偶见IgM、C3在系膜区、血管袢局灶节段沉积。确诊主要依靠电镜及免疫组化检查。电镜下可见系膜区增宽、基质增加、内皮下间隙增宽。可见电子致密的、散在或束状聚集的、直径43~65 nm、规律排列有横纹的纤维状物,在增宽的系膜区、系膜旁区及内皮下积聚。通过免疫组织化学,可明确胶原纤维是单一的Ⅲ型胶原纤维。
本病必须与以下疾病进行鉴别诊断:1.甲-髌综合征。该病为常染色体隐性遗传,因调节Ⅳ型胶原的蛋白其编码基因出现异常而致病。多儿童期发病,表现为指甲、骨骼发育异常或畸形、眼、肾脏等多系统受累。病理上肾小球也有大量胶原纤维沉积,但为Ⅰ型、Ⅲ型、Ⅳ型胶原混杂存在,主要沉积在毛细血管基底膜的致密层。2.纤维样肾小球病。临床表现与胶原Ⅲ肾病相似。病理表现多样,可表现为膜增生性、膜性、系膜增生性,免疫荧光常为IgG、C3强阳性。电镜是诊断的主要依据,可见直径20 nm排列杂乱的纤维样物质沉积,部位不定。3.免疫触须样肾小球病。临床表现与病理特点与胶原Ⅲ肾病类似,区别在于电镜下可见肾小球不同部位及电子致密物中有大量的直径为33~47的微管状纤维。4.膜增生性肾炎。临床表现为持续性低补体血症,其病理特点是系膜细胞、系膜基质中-重度增生,并向内皮下插入,使得基底膜表现为双轨形态。免疫荧光表现为毛细血管袢内皮下较多电子致密物。电镜也有助于鉴别。
对于该病,目前临床上尚无特效的治疗方法。国内外学者曾尝试过激素、免疫抑制(环孢素、环磷酰胺、雷公藤)、控制血压、调脂等治疗,患者蛋白尿等症状多无明显缓解,最终均在数年内进展为终末期肾病,效果均欠佳[5,6,9]。本例患者在给予血管紧张素转化酶抑制剂基础上,加用激素,并曾联合吗替麦考酚酯0.5 g每日2次治疗。追踪复查显示患者尿蛋白无明显缓解,肾功能逐渐缓慢进展,与国内外其余病例对药物的反应相似。
韩辉,魏佳莉.胶原Ⅲ肾小球病1例[DB/OL].中国临床案例成果数据库,2023(2023-01-21).http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2023.e00331.
所有作者均声明本研究不存在利益冲突

























