
患儿,女性,17岁,为先证者。患儿弟弟7岁,与姐姐出现共同临床症状如黑棘皮症、眼球震颤和视力减退为主诉来诊要求治疗。
患儿出生后出现眼球震颤、畏光,随着年龄增长逐渐出现黑棘皮症、肥胖、失明、听力下降、心力衰竭、肾功能衰竭、高血压和肝功能损伤。患儿弟弟7岁,出生后亦逐渐出现眼球震颤、畏光、视力减退和黑棘皮症。
通过对临床症状进行分析和全外显子组测序方法进行高通量测序分析ALMS1基因确诊为Alstrom综合征。
缺少有效治疗方法。
Alstrom综合征是一种罕见的常染色体隐性遗传病,常累及多脏器损伤,通过基因检测确诊,缺少有效治疗方法,临床预后较差。
儿科;肾内科;内分泌科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
Alstrom综合征(Alstrom syndrome,AS)又称肥胖-视网膜变性-糖尿病综合征,是ALMS1基因突变导致的一种罕见、累及多系统的常染色体隐性遗传病[1,2]。1959年由Alstrom等首次报道[3],目前全世界大约1 200例患者确诊AS[4]。本病多见近亲婚配,发病无性别差异,人群发病率低,约为1/100万~9/100万[5,6]。不同年龄AS患者临床症状多样,主要临床特点包括视网膜变性、失明、听力丧失、肝功能损伤、肾功能损伤、2型糖尿病、扩张性心肌病、高血压和多器官功能衰竭等。AS临床缺少有效治疗方法,患者基本死于心力衰竭、肾功能衰竭等多脏器衰竭,平均寿命不超过50岁[2],预后较差。本文分析乌鲁木齐市儿童医院心肾科收治的1名伴有黑棘皮症女性患儿的临床病史,以及弟弟与姐姐共同临床症状如黑棘皮症、眼球震颤和视力减退,通过基因检查确诊Alstrom综合征,结合相关文献,探讨AS主要临床特点、诊断、治疗和预后,进一步加深临床医师对Alstrom综合征罕见病的认识。
患儿,女性,姐姐,17岁,第1胎第1产;弟弟,7岁,第2胎第2产;姐弟俩智力发育正常,父母非近亲婚配,否认遗传代谢性疾病等家族史。姐姐住院期间的临床症状主要为呼吸困难、乏力、端坐呼吸和尿量少。
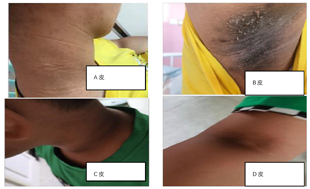
体格检查:呼吸频率40次/min,脉搏120次/min,体质量62 kg,身高165 cm,血压150/110 mmHg,体质指数23.9 kg/m2。神志清,精神差,智力发育正常,呼吸促,颈部、腋下、肘窝处可见皮肤颜色变深,呈灰黑色,皮肤增厚伴有乳突状赘生物。眼睑无浮肿,双肺呼吸音清,心音低钝,律齐,心率120次/min,心前区各瓣膜听诊区无杂音。腹软,肝肋下3 cm,脾未触及肿大,移动性浊音阴性,双下肢轻微浮肿,无多指/趾畸形。
实验室指标:肾功能尿素氮(BUN)18.2 mmol/L,肌酐(CREA)471.6 ummol/L,尿酸(UA)577.1 ummol/L,胱抑素C(Cys C)2.24 mg/L;肝功能丙氨酸转氨酶(ALT)208.4 U/L,天冬氨酸转氨酶(AST)212.1 U/L,白蛋白34.5 g/L;尿蛋白2+,尿微量白蛋白616 mg/L,血糖6.57 mmol/L,尿葡萄糖+;N末端B型脑钠肽>35 000;肾小球滤过率17.14 ml/(min·1.73 m2)。影像学指标:肺CT右肺中上叶及左肺上叶少许炎性变,心影大。心脏彩超左房、左室增大并左心功能不全,二尖瓣关闭不全(中度),三尖瓣关闭不全(轻-中度)并肺动脉高压(轻-中度),主动脉瓣关闭不全(微量),左心室肥厚,心包腔积液。射血分数EF38%,左心室短轴缩短率FS19%。泌尿系B超双肾形态较同龄儿小,实质回声毛糙伴皮髓质界限不清。
住院期间询问病史过程中,母诉患儿弟弟7岁(未住院)与姐姐有相同的黑棘皮症,出生后逐渐出现眼球震颤、畏光和视力变差。姐姐和弟弟临床症状及首发年龄(表1)。
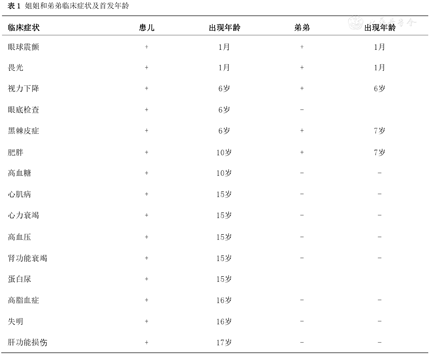
姐姐和弟弟临床症状及首发年龄
姐姐和弟弟临床症状及首发年龄
| 临床症状 | 患儿 | 出现年龄 | 弟弟 | 出现年龄 |
|---|---|---|---|---|
| 眼球震颤 | + | 1月 | + | 1月 |
| 畏光 | + | 1月 | + | 1月 |
| 视力下降 | + | 6岁 | + | 6岁 |
| 眼底检查 | + | 6岁 | - | |
| 黑棘皮症 | + | 6岁 | + | 7岁 |
| 肥胖 | + | 10岁 | + | 7岁 |
| 高血糖 | + | 10岁 | - | - |
| 心肌病 | + | 15岁 | - | - |
| 心力衰竭 | + | 15岁 | - | - |
| 高血压 | + | 15岁 | - | - |
| 肾功能衰竭 | + | 15岁 | - | - |
| 蛋白尿 | + | 15岁 | ||
| 高脂血症 | + | 16岁 | - | - |
| 失明 | + | 16岁 | - | - |
| 肝功能损伤 | + | 17岁 | - | - |
根据姐姐的临床表型(图1),结合姐弟同时患有黑棘皮症(图2)、眼球震颤和畏光,不能除外遗传代谢性疾病,获得家长知情同意,采用全外显子组测序方法进行高通量测序基因检查。
基因检测发现先证者姐姐和弟弟均存在ALMS1基因杂合变异,为外显子8上的突变(图3),其父母分布为ALMS1基因携带者。
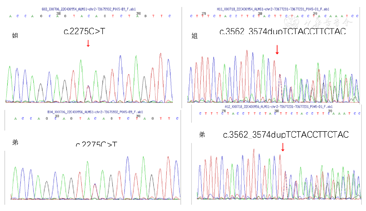
①c.2275C>T,p.Q759X,无义突变,杂合,致病性变异。根据美国医学遗传学会(American College of Medical Genetics,ACMG)指南[7],该变异判定为致病性变异(Pathogenic)PVS1+PM2 Supporting+PM3 Strong:PVS1该变异为零效变异(无义突变),可能导致基因功能丧失;PM2_Supporting在正常人群数据库中频率为0.000 004;PM3 Strong文献数据库已有该位点(Alstrom syndrome)隐性遗传的病例报道,变异标签为DM(致病突变),ClinVar数据库无该位点致病性分析结果。家系分析,先证者父亲该位点杂合变异(图4),先证者母亲该位点无变异,先证者弟弟该位点杂合变异。
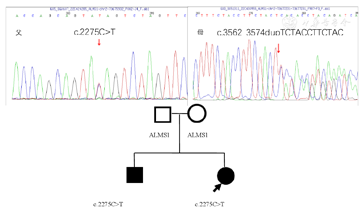
②c.3562_3574dupTCTACCTTCTACT,p.S1192Ffs*21,移码突变,杂合,致病性变异。根据ACMG指南,该变异判定为致病性变异(Pathogenic)PVS1+PVS2 Supporting+PM3(Trans):PVS1该变异为零效变异(移码突变),可能导致基因功能丧失;PM2 Supporting在正常人群数据库中频率为-;PM3(Trans)隐性遗传病,与另一个致病疑似致病变异反式存在(与另一个致病疑似致病突变组成复合杂合)。文献数据库未有该位点相关性报道,ClinVar数据库无该位点致病性分析结果。家系分析,先证者父亲该位点无变异,先证者母亲该位点杂合变异(图4),先证者弟弟该位点杂合变异。
Alstrom综合征是一种常染色体隐性遗传疾病,常累及多器官和多系统,一般在婴幼儿期或幼年期发病,但也有晚期发病的病例。主要临床特征包括视力减退、失明、神经性耳聋、肥胖、扩张型心肌病、2型糖尿病、尿崩症、性腺功能低下、高血压、身体矮小、黑棘皮症等,严重进展为肝、肾、肺部器官损伤并危及生命[8,9]。
AS为单基因遗传疾病,研究证实由ALMS1基因突变导致[10]。ALMS1位于染色体2p13,主要包含23个外显子,编码为4 169个氨基酸组成的相对分子质量约为461.2蛋白质[8]。ALMS1蛋白位于细胞质、细胞骨架、微管组织中心、细胞中心体和纤毛基底部[11,12],在中枢神经系统、循环系统、内分泌系统、光感受器、泌尿生殖系统等多种组织中表达,与纤毛形成有关,参与维持纤毛细胞功能及结构稳定、纤毛细胞信号通路调节、细胞内物质运输、细胞周期调控以及细胞分化[13]。因此,确认AS是与纤毛功能障碍相关的一类遗传疾病[14]。
迄今为止,AS多器官损伤的精确分子机制还没有完全阐明。目前发现约300种不同种族群体的ALMS1基因突变,包括无义突变、插入缺失、移码突变及剪接位点突变、框架位移等[15]。大部分突变发生在ALMS1基因外显子8、10和16,突变概率分别为25%、27%、41%[16,17]。少数突变发生在外显子9、11、12、15、18和内含子17[18,19]。位于外显子8、16的突变,临床表型复杂、病情严重,心脏、肝脏、肾脏、眼、耳等多器官病变,伴有糖类、脂类代谢异常[20]。先证者姐姐的ALMS1基因突变发生在外显子8,临床表型包括肾功能衰竭、心功能衰竭、高血压和失明,病情严重程度与文献报道相符[20]。
Alstrom综合征临床表现复杂多样,不同个体、不同年龄出现不同临床症状,涉及多系统损伤。视力损伤是所有患者生后几周出现最早和公认的症状[21],其他与AS存在重叠临床表现的疾病需要进行鉴别诊断[22]:①.Bardet-Biedl综合征(Bardet-Biedl syndrome,BBS)就是以纤毛病变为基础的一种罕见常染色体隐性遗传病,由于BBS相关基因功能丧失型突变引起非运动性初级纤毛功能障碍,累及全身多个器官系统。主要症状为视网膜变性、肥胖症、多指/趾畸形,性腺发育异常,智力发育迟缓及肾功异常。不同基因突变引起的BBS患者临床表现多样,轻重程度不一,病程呈进行性,致残、致死率较高。AS临床没有多指/趾畸形和智力发育迟缓,可与BBS进行鉴别诊断。②.Laber先天性黑矇(Leber congenital amaurosis,LCA)一种婴儿期起病,表型严重的视网膜营养不良疾病,遗传方式为常染色体隐性遗传,其主要临床表现为视力下降、眼球震颤和严重的视网膜功能障碍,视网膜电流图呈熄灭型的遗传性视网膜疾病。AS视网膜电流图正常。③.Laurence-Moon综合征由PNPLA6基因突变突变导致,主要临床症状包括肥胖、智力低下、视网膜病变、性腺发育不良、脉络膜病变、眼球震颤、痉挛性截瘫、共济失调,无多指或趾。AS智力发育正常,无痉挛性截瘫和共济失调。④.Joubert综合征是包括CEP290等至少36个基因突变,主要症状为视网膜色素变性、多指或趾、肾脏及肝脏异常。有特征性三联征:头颅MRI特异性改变、肌张力减退及发育迟缓,随年龄改善的呼吸异常,眼睛运动异常较常见;泌尿生殖系统异常较常见。AS没有多指或趾和特殊性三联征。⑤.早期出现扩张型心肌病和充血性心力衰竭的AS患儿需要鉴别诊断的疾病为扩张型心肌病、感染性心肌炎。临床进一步通过体质指数、血压、性激素测定、心电图、心脏彩超、视敏度、视网膜电流图、听力测定、尿液分析、肝肾功能、心肌酶、血糖和糖化血红蛋白等一系列检查,评估是否存在肥胖、心血管、肾脏和泌尿系统、内分泌、肝脏、感官系统等异常临床症状。
AS的临床症状和体征会随着年龄出现不同变化。婴儿期出现眼球震颤、畏光、视力下降,囊下白内障、色素性视网膜病,最终进展为失明[23]。儿童期为躯体肥胖、感觉神经性耳聋、高胰岛素血症、胰岛素抵抗,约1/3患儿出现黑棘皮症。青春期表现为首发或复发性扩张性心肌病、2型糖尿病、肾功能衰竭、高血压、身材矮小、高甘油三酯血症、月经不调症、甲状腺功能减低症、男性性腺功能减退、女性高雄激素血症、肾囊肿、反复泌尿道感染、尿崩症和肝硬化等。约60%患儿出生数月内以扩张性心肌病及充血性心力衰竭为首发表现,青春期或成年后会突然复发[24],最终出现心、肾、肝、肺、胰腺和性腺等多器官损伤。姐姐随病情进展出现慢性肾功能衰竭、心力衰竭和高血压多系统损伤,终末期肾脏疾病是AS最常见死亡原因。此外,AS患者伴有睾丸纤维化、男子女性型乳房、运动语言发育迟缓、阵挛性抽搐、反复呼吸道感染、慢性哮喘、肝脏肿大、冠状动脉粥样硬化、脊柱侧弯、脊柱后突凸畸形、脱发和扁平足等症状[2,25,26]。
本文姐弟俩共同症状:婴儿期出现眼球震颤、畏光;儿童期出现黑棘皮症。先证者姐姐青春期逐渐进展为失明、听力下降,临床表型有扩张型心肌病、充血性心力衰竭、肾功能损伤、肝功能损伤、肥胖和高血压。高通量基因测序证实弟弟存在与先证者姐姐相同的ALMS1基因变异,又分别来自父母双方2个ALMS1基因突变,故符合Alstrom综合征诊断。HGMD数据库、ExAC数据库及Clinvar数据库中未收录c.3562_3574dupTCTACCTTCTACT突变位点,拓展了ALMS1基因的表型和突变谱,为ALMS1致病机制以及基因型和表型的关联研究提供了新信息。
Alstrom综合征目前无特异性和根治性治疗手段,预后较差。临床根据不同症状制定个体化、针对性给药方案。Marshall等[27]认为肾脏和胰腺双移植可以作为AS治疗方式;Goerler等通过心肺移植治疗伴有双心室心力衰竭与继发性肺动脉高压的AS综合征[28];Poli经过肾移植手术治疗1例AS合并慢性肾脏病V期的患者,都为AS患者对症治疗提供了思路[29]。基因治疗技术在恢复纤毛功能方面取得成功[30],研究报道PBI-4050是一种相对分子质量为228.3Da的3-戊基苯乙酸钠盐,目前已经在动物实验体外模型中发现,可调节纤维细胞、巨噬细胞、上皮细胞等多种细胞,具有抗炎、预防或逆转纤维化作用,发现在肾脏、心脏、肺和肝脏几个关键器官的抗纤维化活性[31,32]。随着对AS的研究深入,期待着能够为AS临床治疗提供更多有效药物和治疗方法。
Alstrom综合征是罕见病,临床症状复杂多样,基因测序是确诊金标准。作为一种隐性遗传模式,当有分别来自父母双方2个ALMS1基因突变时即可确诊。如果先证者父母均为携带者,子代25%概率患AS、50%是携带者、25%完全正常。产前诊断和植入前诊断有助于优生优育。迄今为止,文献中没有关于AS患者女性怀孕或男性生育的信息[33]。每一个有先证者的AS家庭均应该进行遗传咨询,了解家族发病情况和该疾病病因、遗传风险等,指导再生育。
本文通过对一个AS家系进行高通量测序,证实一对姐弟俩同时存在ALMS1基因突变,比较少见。基因检测发现了ALMS1基因的1个新的突变位点,丰富了AS基因表型和突变谱,为基因治疗研究积累基础。Alstrom综合征早期诊断能够有效评估病情,多学科联合治疗有助于提高患病儿童的诊治水平,更好地改善预后,提高生活质量。
俞蕾.伴有黑棘皮症合并Alstrom综合征的姐弟分析[DB/OL].中国临床案例成果数据库,2023(2023-02-07).http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2023.e00439.
所有作者均声明本研究不存在利益冲突

























