
先证者,社会性别女,2个月22 d,生后发现外生殖器异常病史。
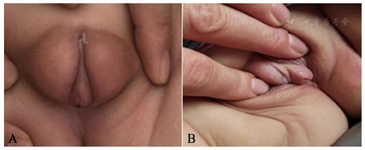
患儿生后外生殖器异常,生长速率正常,无呕吐、腹泻、抽搐等。至当地医院完善性腺彩超可见睾丸,未见子宫、卵巢。体格检查:外生殖器呈女性外观,大阴唇肥厚松弛似阴囊,未触及包块,阴蒂肥大,2.0 cm×1.0 cm,尿道和阴道为同一开口,外生殖器EMS男性化评分:2分,阴毛Tanner 1期。
染色体核型和性染色体FISH结果提示XXY,提示克莱恩费尔特综合征。二代测序提示AR基因杂合致病变异,结合临床考虑合并部分型雄激素不敏感综合征。
择期手术治疗并激素替代治疗。
随访6个月,患儿外生殖器较前无明显变化。
儿科;内分泌科;遗传代谢科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
克莱恩费尔特综合征(Klinefelter syndrome,KS)简称克氏综合征,是男性最常见的性染色体异常疾病,是由于遗传自父方或(和)母方的一条或多条额外X染色体所致,性染色体核型为47,XXY[1]。克氏综合征临床表现有高度异质性,多数患者为正常男性外观。雄激素不敏感综合征(androgen insensitivity syndrome,AIS),是由于Xq12上雄激素受体(androgen receptor,AR)基因突变所致的性发育异常,是46,XY性发育异常的最常见病因[2]。AIS表型变异大,从女性外生殖器到轻度的性发育异常均可见。克氏综合征合并雄激素不敏感综合征的患儿目前国际上报道少,现报道1例克氏综合征合并部分型AIS病例,对其临床诊疗及遗传学特点进行了分析,以期提高临床医生对于该病的认识,减少误诊和漏诊。
患儿,社会性别女,2个月22 d,以"发现外生殖器异常2个月22 d"为代主诉入院。患儿生后发现外生殖器异常,表现为大阴唇肥厚,阴蒂大,排尿位置不详,无排尿时哭闹,无听力障碍,无呕吐、腹泻、抽搐等。病初未在意,大阴唇及阴蒂较前无明显变化。1个月前至当地医院完善性腺彩超可见睾丸,未见子宫、卵巢。为进一步诊治来诊。G3P3,二代试管婴儿,孕期行无创DNA检查提示"性染色体高风险"(未见报告单)。38+3周剖宫产,出生体重3.08 kg。喂养史、生长史无特殊。
母孕早期因"试管婴儿"口服"黄体酮、地屈孕酮、戊酸雌二醇"保胎治疗,孕中后期体健。有2个姐姐(均为自然受孕),"大姐"15岁,来院体检,未行经,Ht 169.2 cm(+1.7 SD)乳房B4,外生殖器幼稚型女性外观,阴道口可明视,阴毛Tanner 1期,无腋毛;当地彩超检查未见子宫及卵巢,彩超腹股沟管内环口可见类似睾丸回声。患儿二姐10岁,乳房B2,外生殖器正常女性外观,阴道口可明视,阴毛Tanner 1期;子宫卵巢彩超正常。
体格检查:体温36.6℃,脉搏112次/min,呼吸26次/min,血压80/50 mmHg(1 mmHg=0.133 kPa),身长59 cm(-0.6 SD),体重6.6 kg(+1 SD)。神志清,精神好,身材匀称,全身皮肤黏膜无色素沉着。头颅无畸形,头围39 cm,前囟直径2 cm×2 cm。心肺腹及神经系统体格检查未见明显异常。外生殖器呈女性外观,大阴唇肥厚松弛似阴囊,未触及包块,阴蒂肥大,2.0 cm×1.0 cm,尿道和阴道为同一开口,外生殖器EMS男性化评分2分,阴毛Tanner 1期(图1)。
血尿便常规正常;肝肾功能心肌酶电解质正常;甲状腺功能正常;8点促肾上腺皮质激素37.31 pg/ml(正常范围:7.2~63.3 pg/ml,以下同);8点皮质醇10.31 μg/dl(6.7~22.6 μg/dl);17α-羟基孕酮2.694 ng/ml(0.31~2.01 ng/ml);抗苗勒氏管激素(AMH)91.17 ng/ml(男55.37~439.45 ng/ml,女0.18~8.26 ng/ml);抑制素B 539.23 pg/ml(男21.00~602.34 pg/ml,女0~226.32 pg/ml);性激素:黄体生成素(LH)1.83 IU/L(男0.10~2.91 IU/L,女0.10~4.01 IU/L);卵泡刺激素(FSH)1.59 IU/L(男0.35~6.94 IU/L,女0.82~7.14 IU/L);雌二醇(E2)<5.00 pg/ml(男0~60.7 pg/ml,女0~398 pg/ml),孕酮(III)0.308 ng/ml(0.03~1.13 ng/ml),睾酮(T)1.03 ng/ml(男0~8.8 ng/ml,女0.35~2.60 ng/ml),双氢睾酮208.3 pg/ml(男40.5~355.0 pg/ml,女23.5~116.0 pg/ml),睾酮/双氢睾酮4.9;硫酸脱氢表雄酮31.28 μg/dl(3.4~124 μg/dl),雄烯二酮0.5 nmol/L(≤2.72 nmol/L)。彩超:膀胱后方未见典型子宫卵巢声像图,会阴部探查未见典型阴道结构回声。双侧睾丸位于腹股沟,形态未见明显异常,左侧大小约10.7 mm×5.7 mm(约0.4 ml),右侧大小约11.7 mm×6.7 mm(约0.6 ml),表面光滑,实质回声分布均匀。肝脾肾彩超:未见明显异常。
患儿"大姐"性激素:LH 30.5 IU/L,FSH 8.2 IU/L,T 5.07 ng/mL(男0~8.8 ng/ml,女0.35~2.60 ng/ml),E2 7.37 pg/ml(男0~60.7 pg/ml,女0~398.0 pg/ml)。
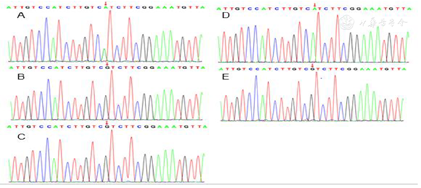
患者外周血染色体核型47,XXY(图2);性染色体荧光原位杂交(FISH)为XXY(图3A);SRY基因荧光原位杂交阳性(图3B)。患儿"大姐"外周染色体核型为46,XY。结合患儿外周血染色体47,XXY及染色体FISH:XXY,诊断克氏综合征。克氏综合征不能解释患儿全部表型,且患儿大姐性发育异常,考虑存在AR基因变异可能,进一步完善全外显子基因检测协诊。


采集患儿及父母、患儿大姐、二姐外周血各2 ml,EDTA抗凝,从受检者样本中提取基因组DNA,构建基因组文库。通过探针杂交捕获目标基因外显子及毗邻剪接区域,并进行富集。对富集的基因进行质量控制,利用高通量测序仪进行全外显子测序。测序原始数据去除不符合质控要求的reads,然后运用BWA软件与UCSC提供的hg19版本人类基因组参考序列进行比对,经过GATK的HaplotypeCaller找出SNV和InDel变异,并对目标区域的覆盖度和测序质量进行评估。依据严格的筛选标准对变异进行过滤,对明确或可能与受检者临床表型相关的基因变异,采用Sanger测序进行验证。对于疑似变异,参照美国医学遗传学和基因组学会(American College of Medical Genetics and Genomics,ACMG)评级指南[3]对其致病性进行评估。
结果提示患儿AR基因第3外显子检测到1个杂合c.1847G>A变异,导致氨基酸改变p.R616H。该变异在人群频率数据库中未收录,HGMD数据库已有该位点的相关性报道,Clinvar数据库该位点的致病性分析为致病。经家系验证分析,受检人父亲、母亲、二姐该位点均无变异,先证者大姐为AR基因c.1847G>A半合子变异。根据ACMG指南,判断为致病(PS2+PM2+PP1+PP3+PP4)(图4)。
本病需与如下疾病鉴别:1.17α羟化酶缺乏症(17α-hydroxylase deficiency,17-OHD):为CYPl7A1基因变异所致常染色体隐性遗传病。17α羟化酶同时具有17α羟化酶和17,20-碳链裂解酶活性。根据17α羟化酶缺乏的类型和严重程度,17-OHD可分为完全型17-OHD、部分型17-OHD。完全型17-OHD患者17α羟化酶活性和17,20-碳链裂解酶活性完全丧失,糖皮质激素及雄雌激素生成障碍,孕酮底物堆积引起盐皮质激素生成过多,临床表现为46XY患者完全女性表型、46XX患者性发育不良,高血压,实验室检查低血钾,孕酮、促肾上腺皮质激素升高,皮质醇降低,同时高促性腺激素性发育不良,基因检测可见CYP17A1基因纯合或复合杂合变异。部分型临床表现相对轻,46XY患者可表现为两性畸形。该患儿支持点:外生殖器异常,不支持点:患儿血压正常,无电解质紊乱、无孕酮、促肾上腺皮质激素、皮质醇水平异常,结合染色体异常及AR基因杂合变异,可排除。2.5α还原酶2缺陷症:为SRD5A2基因变异所致的常染色体隐性遗传病。SRD5A2基因变异,其编码的5α还原酶2活性完全或部分缺失,睾酮不能转化为与雄激素结合能力更强、生物活性更高的双氢睾酮,可引起外生殖器分化障碍,46XY患者可出现小阴茎、尿道下裂、前列腺发育不良等异常,实验室检查睾酮与双氢睾酮比值升高,多大于10,基因检测可见SRD5A2基因纯合或复合杂合变异。该患儿支持点:外生殖器异常,不支持点:患儿性染色体异常,睾丸/双氢睾酮比值正常,结合基因结果,不支持。
联合内分泌、心理及泌尿外科多学科会诊,结合家长意见,继续门诊随访,青春期前确定手术方案,手术前需经医院伦理委员会批准。患儿大姐建议择期行睾丸切除术,术后激素替代治疗,同时进行心理咨询。
目前随访6个月,患儿外生殖器较前无明显变化。
克氏综合征又称先天性曲细精管发育不全、先天性睾丸发育不全综合征,1942年由麻省总医院Klinefelter首次描述,在男性新生儿的患病率为1/660~1/600[1]。雄激素不敏感综合征(androgen insensitivity syndrome,AIS)是由John Morris于1953年首次描述,在男性新生儿中的发病率为1/64 000~1/20 000,为X连锁疾病[4]。AR基因变异及多态可对46XY个体性发育产生不同程度的影响。对于47,XXY的个体,AR基因变异亦可能导致不同程度的性发育异常,在成人克氏综合征正常男性表型的患者研究中发现,AR基因杂合变异在克氏综合征中并不少见,发病率可达5.9%[5]。但因外观性发育异常就诊的患者中,国内外文献仅见数例基因明确的KS合并AIS的患者[6,7,8]。本文中47,XXY同时合并PAIS患者临床罕见。
克氏综合征临床表现有高度的差异性,受多种因素影响,如X染色体数目、AR基因CAG重复序列多态性等,但因Y染色体存在,表型为男性,多数患儿为正常男性外观,性腺为睾丸,部分可表现为小阴茎,尿道下裂少见[1]。AR基因半合子变异可导致雄激素不敏感而出现不同程度的性发育异常,根据患儿生殖器的表型,又可将AIS分为完全型(complete AIS,CAIS)、部分型(partial AIS,PAIS)及轻型(mild AIS,MAIS)[9]。CAIS患者具有男性内生殖器和正常的女性外生殖器,表现为盲端阴道、子宫卵巢缺如。PAIS患者表现为不同程度的外生殖器异常,常有小阴茎,尿道下裂,睾丸小,隐睾多见,男性化程度严重不足的患者可表现为重度尿道下裂、阴茎似阴蒂。MAIS患者表现为正常的男性外生殖器,可出现青春期男性乳房发育、不育等。本文中先证者染色体结果为克氏综合征,但临床表型为严重的男性化不足,与克氏综合征临床表现不完全相符,故进一步完善检查诊断为克氏综合征合并部分型雄激素不敏感。
性激素方面,本患儿LH、FSH及T水平符合同年龄儿小青春期水平。既往研究发现青春期启动前,KS及AIS患者血清LH、FSH、T的基础水平与同龄儿童相比无显著差异,青春期启动后KS患者T最终下降至低于正常范围,LH、FSH逐渐升高[1]。AIS患者T及E2处于男性正常高限或升高,LH常高于正常男性,FSH与正常男性水平相同或升高。目前KS合并AIS患者性激素水平情况尚不清楚。成人正常男性外生殖器KS患者中存在AR基因杂合变异与无变异患者相比,性激素水平无明显差异[5]。目前国内外共报道4例因性发育异常就诊且明确的KS合并AIS患者,确诊年龄1.1~30岁,2例因外生殖器异常就诊,2例因原发性闭经就诊[6,7,8,10]。性激素水平方面,幼儿期患者性激素水平符合同龄儿童,青春期患者可见LH及FSH明显升高,性激素水平符合KS患者水平,报道中11岁患者T水平高于文献中正常值,虽T水平升高,但LH水平仍高,可能与雄激素抵抗有关[2,6]。
AR基因由8个外显子组成,编码920个氨基酸残基的蛋白质。迄今为止,已发现的可导致雄激素不敏感综合征的AR基因突变超过1000种[11]。主要类型包括基因缺失、插入和错义突变。AR蛋白是由4个功能域组成的单链多肽,分别为:①N端结构域(NTD);②DNA结合结构域(DBD);③铰链结构域;④C端配体结合结构域(LBD)。DBD区由外显子2和3编码,由两组锌指蛋白组成,一种与特异识别激素反应原件有关,另一种对稳定DNA受体蛋白有重要作用。该患儿位点位于DBD区,既往在一对表型为PAIS的同胞中被报道[12]。体外功能试验提示突变后AR蛋白无功能。本例中先证者大姐16岁未行经,外生殖器体格检查为正常女性外生殖器,无阴毛及腋毛,进一步完善检查为46,XY完全型雄激素不敏感,与既往报道相符。先证者X染色体杂合变异,但出现严重外生殖器男性化不足表现,推测与先证者正常X染色体失活或突变染色体优势表达有关。既往文献报道中因性发育异常就诊且确诊为KS合并AIS的患者中,CAIS者共3例,为KS合并AR基因纯合及复合杂合变异,PAIS者共1例,为AR基因杂合变异[6,7,8,10]。本例患儿表现为PAIS,亦为AR基因杂合变异。其他报道AR基因外显子1的CAG重复序列多态性等亦可能影响患儿表型,目前该患儿未完善。该先证者及大姐均出现AR基因变异,但父母均正常,需考虑生殖细胞嵌合可能,再生育需注意遗传咨询。
KS合并PAIS尚无治疗经验[10]。虽PAIS青春期可能出现阴蒂进一步增大,但因患儿合并KS,考虑性腺功能可能进行性衰退,进行性别选择时需注意参考。因PAIS可能对患儿心理产生影响以及青春期PAIS对性发育的影响,一般建议如选择女性需尽早手术治疗,目前患儿门诊随访中,注意定期性腺彩超检查观察有无恶变可能。CAIS一般选择女性身份,因Y染色体的存在,睾丸发生生殖细胞肿瘤风险增加,但因青春期前睾丸发生癌变概率低,且雄激素可通过体内芳香化酶的作用转化为雌激素作用于正常的雌激素受体,对患儿骨密度、青春期生长及女性第二性征发育有重要作用,故有学者建议青春期后手术切除性腺,同时建议进行激素替代及心理干预[13]。先证者大姐目前诊断CAIS,现已青春期后期,建议可手术探查睾丸并行手术切除。
综上所述,本文描述了1例克氏综合征合并部分型雄激素不敏感的患者。正常X染色体的随机失活可能是克氏综合征患儿出现部分型雄激素不敏感的病因,提示对于克氏综合征患者,出现严重男性化不足时需注意合并雄激素不敏感可能。
陈琼,吴雪,毋盛楠,等.47,XXY并部分型雄激素不敏感综合征1例[DB/OL].中国临床案例成果数据库,2023(2023-02-09).http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2023.e00475.
所有作者均声明本研究不存在利益冲突

























