
患者,女性,27岁,孕4产1,以"宫内孕29周,超声发现羊水过多1个月余"为主诉来院就诊。孕期定期围产保健,早孕期胎儿颈项透明层测量正常,中孕期血清学筛查低风险,孕22周胎儿超声系统结构筛查及胎儿心脏彩超未提示异常,孕25周外院彩超提示羊水偏多,羊水指数220 cm,OGTT筛查未见异常,孕期无毒物、药物、射线等不良接触,夫妻双方无不良家族史,夫妻双方外观及体格检查未见异常。
妊娠29周胎儿系统超声检查结果提示双顶径81 mm(+2.1 SD,按NICHD亚裔曲线计算,下同),头围279 mm(+1 SD),腹围274 mm(+2.6 SD),股骨长径48 mm(-2.5 SD),胎儿头颅脑中线居中,颅内结构近场远场清晰度相似,近场颅骨光环受压变形,移开探头可恢复后,胎儿鼻骨未显示,胎儿羊水最大深度73 mm,指数203 mm,余胎儿结构未见异常。妊娠32周胎儿针对性超声检查结果提示双顶径91 mm(+2.6 SD),头围310mm(+1.6 SD),腹围317 mm(+3.2 SD),股骨长径58 mm(−0.6 SD),胎儿颅骨顶枕部膨隆,颅骨受压变形,近场颅内结构显示清晰,鼻骨未显示,右侧锁骨长3.84 mm,左侧锁骨未显示。
获取胎儿羊水及父母外周血进行染色体微阵列(CMA)检查及全外显子测序(WES)检测,结果显示胎儿RUNX2基因c.911-914delinsTTT新发杂合变异,双亲Sanger测序均为野生型。RUNX2基因c.911-914delinsTTT变异位点在中国人群中尚未见报道,扩展了我国RUNX2基因的突变谱。
多学科会诊,伦理委员会讨论,充分告知父母出生后该病的临床表现及现有治疗该病的医疗技术方法。
胎儿父母选择终止妊娠。
产科;遗传科;儿科;影像科;产前诊断


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
颅骨锁骨发育不良(cleidocranial dysplasia,CCD,OMIM 119600),是一种罕见常染色体显性遗传的骨骼发育不良疾病,约60%的CCD时是由矮小相关转录因子2基因(RUNX2,OMIM 600211)的变异所致[1,2]。RUNX2是成骨细胞分化和骨骼发育的关键调节因子[2]。不同的RUNX2突变与疾病的严重程度无关。CCD的表型差异很大,按照有无遗传学背景及症状的轻重不同,将该病分为3型:Ⅰ型为标准型,符合Marie及Sainton标准,即有家族遗传背景,颅骨、锁骨、骨盆等均受累;Ⅱ型有家族遗传型,但颅骨不受累;Ⅲ型为散发型,无家族遗传性。无家族遗传背景者容易被误诊。目前,多数CCD因出生后牙齿或骨骼发育异常被报道,其胎儿期表型相关报告较少。本文就1例散发RUNX2基因变异的CCD影像学表型及产前遗传学诊断进行详细的分析,以期为该病的产前诊断及遗传咨询提供帮助。
患者,女性,27岁,以"宫内孕29周,超声发现羊水过多1个月余"为主诉来院就诊。孕妇自然受孕,孕期定期围产保健,早孕期胎儿颈项透明层测量正常,中孕期血清学筛查低风险,孕22周胎儿超声系统结构筛查未提示异常,孕25周外院彩超提示羊水偏多,羊水指数220 cm,OGTT筛查未见异常,孕期无毒物、药物、射线等不良接触。孕29周外院行胎儿系统超声检查提示羊水过多,余胎儿结构未见异常,遂转诊至我院就诊。来院后于我院行胎儿系统彩超检查及胎儿实时三维颅脑针对性超声检查;在患者知情同意并签署知情同意书下,行羊水穿刺术获取的胎儿羊水标本,及抽取夫妇外周血标本,进行染色体微阵列(CMA)检查及全外显子测序(WES)检测。孕妇平素体健,配偶体健,育有1女,现体健,夫妻双方无异常家族史夫,双方外观及体格检查未见异常。
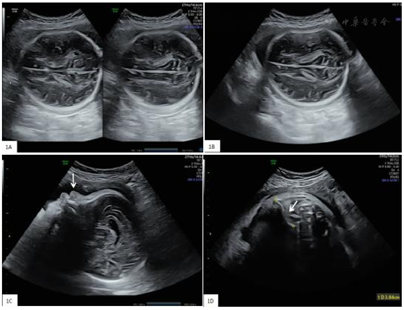
妊娠29周我院胎儿超声检查结果提示双顶径81 mm(+2.1 SD,按NICHD亚裔曲线计算,下同),头围279 mm(+1 SD),腹围274 mm(+2.6 SD),股骨长径48 mm(−2.5 SD),第六脑室存在宽约9.5 mm,胎儿头颅脑中线居中,颅内结构近场远场清晰度相似,近场颅骨光环受压变形,移开探头可恢复后(图1A、图1B)。鼻骨未显示(图1C)。胎儿羊水最大深度73 mm,指数203 mm,余胎儿结构未见异常。妊娠32周我院胎儿超声检查结果提示双顶径91 mm(+2.6 SD),头围310 mm(+1.6 SD),腹围317 mm(+3.2 SD),股骨长径58 mm(−0.6 SD),胎儿颅骨顶枕部膨隆,颅骨受压变形,近场颅内结构显示清晰,鼻骨未显示,右侧锁骨长3.84 mm(图1D),左侧锁骨未显示。
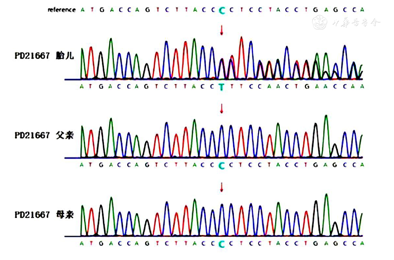
胎儿样本未检测到染色体非整倍体或100 kb以上已知的明确致病的基因组拷贝数变异。胎儿检出RUNX2(OMIM:600211)基因:c.911-914delinsTTT杂合变异,Sanger测序父亲和母亲均为野生型(图2),该变异引起基因开放阅读框发生改变,导致蛋白功能改变(PVS1),在神州基因组数据库、人类外显子数据库(ExAC)、参考人群千人基因组(1000G)和人群基因组变异频率数据库(gnomAD)中没有发现(PM2_Supporting),且该变异为未经双亲亲缘关系验证的新发变异(PM6);根据ACMG(美国医学遗传学与基因组学学会)颁布的遗传变异分类指标与指南,该位点变异评级为致病性(PVS1+PM2_Supporting+PM6),与常染色体显性遗传颅骨锁骨发育不良相关(OMIM:119600)。
根据患者影像学检查及染色体微阵列(CMA)检查及全外显子测序(WES)检测结果,诊断考虑为:1.胎儿RUNX2基因致病变异;2.胎儿颅骨锁骨发育不良;3.胎儿羊水偏多;4.宫内孕32周;5.孕4产1。根据影像学检查结果还需要与常染色体隐性遗传Gillessen-Kaesbach-Nishimura综合征、骨软骨发育不良、成骨不全等相鉴别;这几种疾病均可能有骨化不足的影像表现,但Gillessen-Kaesbach-Nishimura综合征常合并其他结构异常如多囊肾和先天性心脏缺陷,骨软骨发育不良存在四肢长骨短小,成骨不全有长骨弯曲、骨折等表现,通过超声影像学可基本排除,最终鉴别依据基因遗传学检测结果。
患者来院后,完善超声检查后,组织产科、骨科、神经内科、超声科、产前诊断与遗传科进行多学科会诊。患者妊娠孕周较大,在充分知情同意的情况下进行了介入行产前诊断遗传学检测。遗传检测结果提示RUNX2基因致病变异,考虑颅骨锁骨发育不良,再次进行胎儿超声复查并组织遗传学科、伦理委员会、产科、骨科、神经内科、超声科、产前诊断科进行多学科讨论,充分告知父母颅骨锁骨发育不良表型差异很大,从仅有轻度牙齿畸形的患者到严重骨异常的患者,主要表现为全身骨骼和牙齿发育不全,其特有的临床特征为锁骨缺如/发育不全,囟门未闭合/闭合不全,牙齿萌出异常/多生牙等,大多数CCD患者智力正常,罕见的患者会表现出认知缺陷,及现有治疗该病的医疗技术方法及分娩时注意事项,并告知夫妇本次胎儿RUNX2基因致病变异为新发变异,下次妊娠再发风险较低,但不排除生殖腺嵌合可能,建议下次妊娠进行绒毛穿刺,羊水穿刺等介入性产前诊断。
经多学科会诊及伦理委员会讨论,尊重胎儿父母意愿及诉求,在妊娠32周终止妊娠。
颅骨锁骨发育不良(CCD)的患病率为1∶1 000 000,是一种罕见的常染色体显性遗传病,其特征是锁骨发育不全和(或)发育不全、面中部发育不全、颅缝持续开放或延迟闭合、身材中度矮小、恒牙列和多生牙萌出延迟[3]。临床表型差异很大,从仅有轻度牙齿畸形的患者到严重骨异常的患者。即使在一个家庭中,也有相当大的表型变异被报道[4]。X射线提示多个骨骼的受累,包括颅骨、胸部、骨盆、四肢和牙齿异常。大多数CCD患者智力正常,罕见的患者会表现出认知缺陷,Toshiki等[5]认为可能与CCD颅骨骨化延迟,自然分娩时CCD胎儿的大脑受到潜在的损害引起的脑软化有关。因颅骨钙化减少,阴道分娩过程中发生的压力可能导致颅骨过度变形引起新生儿脑实质内出血、后颅窝硬膜下血肿[6,7]。因此,早期识别CCD具有临床意义。
产前超声可以诊断CCD最早发表于1990年代中期[8,9]。产前超声主要征象是胎儿的颅骨骨化差,并伴有短锁骨或锁骨发育不全[10,11]。但这些研究均认为,由于锁骨发育不全的鉴别诊断包括许多骨骼发育不良,因此需要结合胎儿的其他骨骼发现来评估CCD。Soto等[12]研究采用3D超声产前诊断18周3 d的CCD胎儿主要超声表型为鼻骨缺失,并伴有颅骨低钙化。Hermann等[13]认为颅骨钙化不足及鼻骨缺失是CCD患者在出生前和出生后易于识别的特征,而且3D超声有利于识别这些超声影像。目前仅有少数散发CCD的产前超声报道。本文报道1例散发CCD的产前超声表现,主要表现为胎儿颅骨钙化差,顶枕部膨隆,颅骨受压变形,近场颅内结构显示清晰,鼻骨缺失,左侧锁骨不显示。引产后检查发现胎儿颅骨软,左侧锁骨未触及,遗憾的是孕妇拒绝尸检及放射性X线进一步检查。至于羊水过多是否是CCD产前超声的表型,目前尚未见报道,尚需更多的病例研究,但发现羊水过多时,超声检查时更应仔细,以免漏诊。也有文献报道,CCD患者可表现为腭盖高拱、不完全或完全性腭裂、下颌正中联合闭合延迟,这可能影响胎儿的吞咽功能。
RUNX2位于染色体6p21上,有8个编码外显子,是成骨细胞分化和骨骼发育的关键调节因子,能够在转录水平调节成骨相关因子的表达[14],RUNX2基因突变可能会干扰DNA结合活性、改变蛋白的核定位或产生一种生物学上不活跃的突变体或截短蛋白,因此,RUNX2的表达必须严格控制,以保证骨骼的正常发育[15]。在典型颅锁骨发育不全的患者中,大多数RUNX2变异发生在Runt结构域,主要是错义变异,无义变异和移码变异在整个基因中均有检出。发生在Runt结构域内致病变异破坏了蛋白的反式激活活性,从而导致典型的颅锁骨发育不全临床表型,其牙齿发育不良、颅面部异常和矮小身材表型要比发生在Runt结构域外的变异所导致的相应表型更严重[16]。脯氨酸-丝氨酸-苏氨酸富集区(PST功能域)也是突变热点区域,该结构域从5号外显子到8号外显子,包含翻译后修饰的序列,它对反式激活或转录抑制功能很重要,位于PST结构域内的NMTS调节RUNX2与核基质中亚核位置的结合,PST结构域的突变可能会损害RUNX2的反式激活或转录抑制功能[17,19]。有学者报道国内两例典型的CCD患者RUNX2:c.1111dupT及c.1550delT移码突变,两种突变均可导致RUNX2对骨钙素启动子的反式激活活性丧失[19,20]。本文发现RUNX2 c.911-914delinsTTT(p.P304Lfs*4)新的变异,该突变会导致杂合子中PST结构域的丢失,目前这个突变位点尚未见报道,我们将对这个变异位点导致的蛋白功能异常的分子机制进行进一步研究。
随着分子遗传学的发展,WES检测在临床已经广泛使用,是罹患单基因疾病胎儿的重要检测手段。ACMG指南提出对于影像学提示结构发育异常的胎儿,可考虑行WES检测。颅脑发育异常与许多遗传性疾病相关,当胎儿期超声发现颅骨钙化不足时,应由临床经验丰富的超声医生进行胎儿影像学检查,尤其是详细进行其他骨骼系统的检查,若同时发现发育不全的锁骨及鼻骨发育异常,应考虑颅骨锁骨发育不良可能。根据胎儿影像学表型行家系WES及CNV-seq进行遗传学诊断,此方案能够提高异常检出率,缩短检测周期,快速鉴定是否新发变异及双亲基因型,但若孕妇因经济因素等不能接受家系WES及CNV-seq检查时,应充分告知孕妇产前单人WES检测的局限性及弊端,并签署知情同意书。
CCD的诊断需结合影像学检查和遗传学诊断结果。在具有已知突变的家庭中,产前遗传学诊断是首选,但对于无家族史的散发病例,产前超声检查尤为重要,通过对本例散发型颅骨锁骨发育不良胎儿期影像学结果的分析,可以提高临床医生对此病的产前识别,发育不全的锁骨、颅骨钙化不足及鼻骨缺失是产前超声易观察到的主要特征,3D超声更有助于胎儿期的影像学观察。本研究中根据影像学表型,进行基因诊断所确定的RUNX2 c.911-914delinsTTT致病位点突变,在中国人群中尚未见报道,扩展了我国RUNX2基因的突变谱,有助于该病遗传咨询和产前诊断。
所有作者均声明本研究不存在利益冲突
袁莉敏,刘灵,翟闪闪.产前诊断散发型颅骨锁骨发育不良1例[DB/OL].中国临床案例成果数据库,2023(2023-02-22).http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2023.e00718.

























