
患儿,男性,1岁3个月,因"间断抽搐2个月"于2022年1月就诊。
患儿于1岁1个月龄时无明显诱因出现抽搐发作,表现为:双眼凝视上翻,呼之不应,偶伴肢体抖动、双手握拳、双足趾屈、面色发绀,不伴牙关紧闭,持续时间十余秒至3 min,2~7次/d。患儿3个月抬头,6个月会坐,8个月会爬,不会叫"爸爸、妈妈",起病前可独站,可扶走,会拇食指对捏物体,有认生表现。起病后发育倒退,不能独坐,无精细运动,无语言,反应差。查体:身长80 cm(0~1 SD),体重9 kg(−2~−1 SD),头围42.8 cm(<−3 SD),精神欠佳,额部长,眼距宽,双眼内聚。对声音、光线刺激反应差,可四点支撑,不能独坐,不会拇食指对捏,不会说话,四肢肌张力低,肌力3级。
临床表现癫痫发作、全面发育迟缓和特殊面容,结合全外显子组测序检测出SLC9A6基因intron11:c.1366+1G>C新发剪切变异,诊断为Christianson综合征。
给予口服抗癫痫发作药物左乙拉西坦和丙戊酸钠,并给予康复治疗。
癫痫控制;缓慢进步,仍有全面发育迟缓,现患儿2岁4个月龄,可独站,可扶走,不会拇食指对捏物体;仅能无意识发"妈妈"音,语言理解能力差;能和同龄儿童玩耍,但适应能力欠佳,不能执行指令,不会模仿,有认生表现。
儿科;神经内科;内分泌遗传代谢科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
智力发育障碍,X连锁综合征,Christianson型(Mental retardation,X-linked syndromic,Christianson type,MRXSCH,OMIM:300243)又名Christianson综合征(Christianson syndrome,CS),是一种X连锁遗传方式的发育性脑功能障碍性疾病,其临床特征是进行性加重的发育迟缓、发育倒退、智力残疾,还可能涉及缄默症、自闭症症状、颅面部畸形、共济失调、眼肌麻痹、不同类型的早发性癫痫发作以及小脑和脑干萎缩,女性携带者可能会受到轻微影响[1,2,3,4]。编码钠氢交换蛋白成员6(Na+/H+ exchanger protein member 6,NHE6)的SLC9A6(solute carrier family 9 member A6)基因的致病性突变与Christianson综合征和孤独症谱系障碍相关[5]。自1999年Christianson等首次发现并报道至2022年10月,目前国内外报道有99例,其中国内仅有5例报道。本文报道郑州大学第三附属医院诊断的1例以癫痫发作和全面发育迟缓为主要临床表现的1岁3个月龄男童,分析其家系WES检测结果,为X染色体SLC9A6基因intron11:c.1366+1G>C新发剪切变异,该变异在人类外显子数据库(ExAC)、参考人群千人数据库(1000G)和人群基因组突变频率数据库(gnomAD)中均未报道。该患儿存在频发室性早搏,动态心电图可见室性并行心律,而在已报道的文献中并无Christianson综合征患者存在心律失常的描述,此患儿心脏MRI结果显示左心室心尖段及基底段心肌炎改变,推测与心律失常相关,但具体机制仍需进一步研究。该病例报道拓宽了SLC9A6基因突变谱,增加了中国Christianson综合征的临床表型,加深对Christianson综合征的认识。
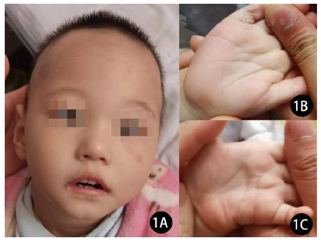
患儿,男性,1岁3个月,因"间断抽搐2个月"于2022年1月至郑州大学第三附属医院就诊。患儿为第3胎,第3产,足月因"胎盘早剥"行剖宫产娩出,新生儿血筛查无异常。患儿3个月抬头,6个月会坐,8个月会爬,不会叫"爸爸,妈妈",起病前可独站,可扶走,会拇食指对捏物体,有认生表现,2个月前(1岁1个月龄)无明显诱因出现抽搐发作,表现为双眼凝视上翻,呼之不应,偶伴肢体抖动、双手握拳、双足趾屈、面色发绀,无牙关紧闭,持续时间十余秒至3 min,2~7次/d。此后出现发育倒退,现可四点支撑,不能独坐,无精细运动,无语言,反应差。体格检查:身长80 cm(0~1 SD),体重9 kg(−2~−1 SD),头围42.8 cm(<−3 SD),前囟已闭,对声音、光线刺激反应差,额部长,眼距宽,双眼内聚,左手通贯掌(图1)。心肺腹无异常,四肢肌力3级,肌张力低,腱反射引出正常,病理征、脑膜刺激征阴性。家族史无特殊,有两个同父异母哥哥,体健。
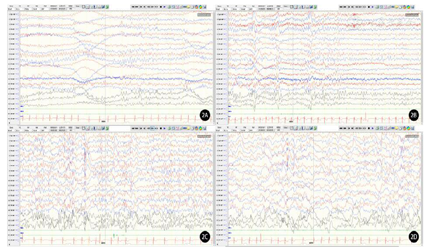
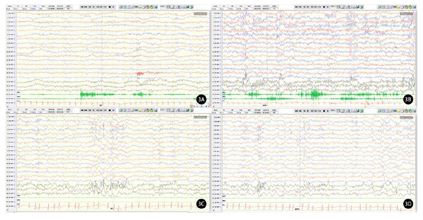
入院后检查:肝肾心功能、电解质、血糖、甲状腺功能、血氨、铜蓝蛋白、乳酸、丙酮酸、血氨基酸及酰基肉碱串联质谱分析、外周血染色体核型等均未见明显异常。四肢肌电图检查正常。Albert婴儿运动量表(2021年12月23日,1岁2个月龄):总分52,百分比<5%。双侧肢体肌4+级,双下肢肌张力低,表情淡漠,目光对视欠佳,会扶家具侧行迈步4步,不会独站;整体印象:该儿童整体发育落后,服从指令欠佳。头颅核磁共振(2021年12月12日,1岁2个月龄):大枕大池,双侧颞角稍宽。动态心电图(2022年1月20日,1岁3个月龄):可见窦性心律不齐;频发室性早搏,室早总数:5678次(3.53%),二联律2阵,三联律266阵。复查动态心电图(2022年5月5日,1岁7个月龄):室早总数5036个,二联律82阵,三联律20阵,可见室性并行心律。心脏彩超(2021年12月1日,1岁2个月龄):心内结构及功能未见明显异常。心脏MRI(2022年5月15日,1岁7个月龄)可见左心室心尖段及基底段心肌炎改变。视频脑电图(video-electroencephalography,VEEG)(2022年1月17日,1岁3个月龄):清醒安静状态下双侧枕区为低-中幅3~5 Hz慢波及少量5~7 Hz θ波,左右基本对称;醒睡各期多量多灶低-高波幅棘波、棘慢、多棘慢、慢波阵发、散发,偶见广泛,睡眠期著(图2)。复查VEEG(2022年4月6日,1岁6个月龄):清醒安静闭目时双侧枕区为低-中幅6~8 Hz α波;醒睡各期双侧额极、额、中央区大量低-高幅棘波、棘慢波散发、阵发或连发,睡眠期著;醒睡各期双侧额极、额、中央、中线区大量低波幅快波活动阵发,清醒期著(图3)。
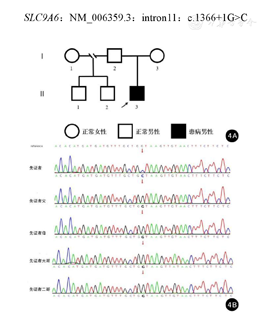
经患儿父母知情同意,并由郑州大学第三附属医院伦理委员会批准(伦理号:2021-062-01),采集患儿及其父母、两个哥哥全血各2 ml。从外周血中提取基因组DNA,进行家系全外显子测序(北京贝瑞和康生物技术有限公司),基因检测结果显示患儿X染色体SLC9A6基因存在新发intron11:c.1366+1G>C剪切突变,先证者父母及两个哥哥均未见突变(图4)。根据2019美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)制定的遗传变异分类标准与指南进行致病性分析:LOF变异导致基因功能可能丧失(PVS1);该变异为家系样本中经双亲验证的新发变异(PS2);该位点在人类外显子数据库(ExAC)、参考人群千人数据库(1000G)和人群基因组突变频率数据库(gnomAD)等均未收录(PM2),此变异评级为致病性变异:PVS1+PS2+PM2。该变异位于11号内含子剪切位点,推测由于剪切突变导致SLC9A6基因剪切异常,影响12号外显子,从而导致疾病发生。
患儿自幼发育迟缓,间断抽搐、发育倒退2个月,体格检查面容特殊,结合基因检测结果,诊断为SLC9A6基因突变导致Christianson综合征;患儿动态心电图频发室性早搏,可见室性并行心律,诊断为室性早搏。
鉴别诊断:(1)Angelman综合征(AS)。AS是一种神经发育障碍性疾病,其特征是智力低下、运动或平衡障碍、典型的异常行为以及言语和语言功能的严重受损。大多数病例是由于缺乏母体对染色体15q11~q13上印记区域的贡献所引起。该患儿存在严重的发育落后、特殊面容、抽搐支持诊断,但患儿出现发育倒退,以及相关遗传学检测可排除该诊断。(2)先天性糖基化障碍(congenital disorder of glycosylation,CDG)。CDG是由蛋白质和脂质的N-糖苷链和O-糖苷链糖基化途径异常引起的一组累及多器官系统的遗传代谢性疾病,该病大部分是常染色体隐性遗传方式。该患儿存在抽搐及发育倒退支持诊断,代谢相关检测正常不支持诊断,相关遗传学检测可排除。(3)脑性瘫痪(cerebral palsy,CP)。CP是一组持续存在的中枢性运动和姿势发育障碍、活动受限症候群,这种症候群是由于发育中的胎儿或婴幼儿脑部非进行性损伤所致。脑瘫的运动障碍常伴有感觉、知觉、认知、交流和行为障碍以及癫痫和继发性肌肉、骨骼等问题。患儿肌张力低、运动功能异常支持诊断,孕期、围产期、新生儿期无异常病史,无姿势异常、锥体束征阳性等特征、波动性病程、起病晚不支持诊断。
患儿2021年11月30日出现抽搐发作,外院加用奥卡西平联合丙戊酸钠治疗,无效,2022年1月就诊于我院,加用左乙拉西坦,渐停奥卡西平;给予营养心肌及功能训练等治疗。
于我院治疗后间隔1个月四肢肌力恢复正常。随访至2023年2月,患儿目前口服丙戊酸钠口服液20 mg/(kg·d),左乙拉西坦口服液38 mg/(kg·d)。现一般情况可,近2个月无抽搐发作,发育缓慢进步。现患儿2岁4个月龄,可独站,可扶走,不会拇示指对捏物体;仅能无意识发"妈妈"音,语言理解能力差;能和同龄儿童玩耍,但适应能力欠佳,不能执行指令,不会模仿,有认生表现,Griffiths神经发育评估(2023年2月21日,2岁5个月龄):运动相当于11个月,个人与社会相当于9个月,语言相当于7.5个月,手眼协调相当于8.5个月,表现相当于10.5个月。
男性发病率高于女性的现象在智力低下人群中早已被认识,且人们认为这与X连锁精神发育迟滞(X-linked mental retardation,XLMR)基因有关。智力发育障碍,X连锁综合征,Christianson型是一种罕见的X连锁隐性遗传的单基因疾病,据统计,其患病率在1∶16 000~1∶100 000,男性患病多见,是由位于X染色体长臂上的SLC9A6基因(位点Xq26.3)变异导致一种与小头畸形、癫痫发作、共济失调和言语缺失相关的智力发育障碍综合征[6,7]。本病患者身材瘦小,10岁后可能无法行走,部分患者还可能有小脑和脑干萎缩,其脑电图具有快(10~14 Hz)的背景节律的特点,可见额极、额、前颞和额中线区的尖波、慢波[5]。2014年Pescosolido等[2]提出了Christianson综合征的诊断标准:核心诊断症状包括男孩儿童早期发病、缄默症、中度至重度智力障碍、癫痫、躯干共济失调、小头畸形以及多动行为。通常出现的继发症状包括自闭症、Angelman综合征症状、眼球运动异常、发育退化,尤其是10岁后失去独立行走能力、消瘦和小脑蚓萎缩。本例患儿的核心症状为癫痫、发育倒退和缄默状态,查体发现特殊面容、肌张力低下,未发现异常行为,结合患儿临床表现、基因检测结果等,诊断为Christianson综合征明确。
1999年Christianson等[8]首次描述了一个患有XLMR的南非家族,其致病基因位于Xq24~q27,该家族由16名患病男性和10名携带者女性组成。所有男性共同临床特征包括智力发育严重受损、缄默症、强直-阵挛发作的癫痫和预期寿命有限。此外还有轻度颅面畸形、眼肌麻痹、小脑和脑干萎缩,10名女性携带者中有3名智力发育轻度受损,存在学习和行为问题。自首次报道后多个国家均进行了病例报道,我们检索并汇总了自1999年至2022年10月Christianson综合征相关文献,近有58个家系,95例患者。所有患者均存在严重的精神运动发育迟缓(95/95,100.00%),其中约半数患者合并发育倒退(25/55,45.45%),余常见的临床表现依次为癫痫(87/88,98.86%)、缄默状态(75/83,90.36%)、小头畸形(61/77,79.22%)、共济失调(49/63,77.78%)、自闭症(35/47,74.47%)、眼球运动障碍(35/49,71.43%)及运动功能亢进(40/56,71.43%),这些患者还可表现为睡眠障碍(17/35,48.57%)、Angelman综合征症状(23/50,46.00%)和小脑萎缩(20/57,35.09%)。
通过汇总文献已报道的病例可知,近98.91%的Christianson综合征患者存在癫痫,脑电图背景节律可为正常或异常快波,发作间期癫痫样放电可为局灶、多灶或广泛[9]。本例患儿脑电图背景节律正常,存在以前头部、中线为主的棘波、棘慢波,与已报道病例相符。对详细报道癫痫相关情况的55例患者临床表现进行分析,我们发现Christianson综合征患者的癫痫发作多起始于婴幼儿时期(均在3岁内起病),几乎所有病例均存在强直阵挛发作,且以睡眠期发作为主;尽管使用多种抗癫痫发作药物治疗仍控制不佳,据报道丙戊酸钠、左乙拉西坦、氯硝西泮及拉莫三嗪使用较多,且对癫痫发作的控制更加有效;部分病例存在癫痫丛集性发作后出现发育倒退的现象[1,2,10,11]。2020年Ikeda等[11]报道的1例患儿于3岁时对抗癫痫发作药物耐药后出现发育倒退,由此我们推测此现象可能是由于无法控制的频繁癫痫发作所引起的患儿进行性脑功能障碍。本文报道的患儿属于急性起病、波动性病程,丛集性癫痫发作,起病前发育落后,表现为不会叫"爸爸、妈妈",可扶走;起病后发育倒退,表现为不能独坐、爬、独站。既往曾使用苯巴比妥、地西泮止痉及口服奥卡西平抗癫痫发作,效果欠佳,目前口服丙戊酸钠、左乙拉西坦治疗,抽搐控制,脑电图间期仍存异常放电。
Christianson综合征近34.48%的患者MRI表现为小脑萎缩,主要影响半球和蚓部,双侧颞叶、枕叶蛛网膜下腔增宽共见16次报道,可能是由于皮质发育不良或萎缩引起。本例患儿于1岁2个月龄行头颅MRI示双侧颞角稍宽,未见明显异常,有待继续随访观察相关情况。此外,我们报道此例患儿存在频发室性早搏,动态心电图可见室性并行心律,在已报道的文献中并无Christianson综合征患者存在心律失常的描述,此患儿心脏MRI结果显示左心室心尖段及基底段心肌炎改变,推测与心律失常相关,但具体机制仍需进一步研究。
2008年Gilfillan等[7]对其报道的挪威家系在Xq24~q27.3区间进行测序分析,发现SLC9A6基因编码区存在6个碱基的缺失,位于Xq26.3区,这一缺失导致该基因所编码的NHE6多肽的Na+/H+交换域失去了两个高度保守的氨基酸残基(p.E255_S256 del),这些残基已被证明对离子的迁移是必不可少的,且这种序列变异并非多态性。汇总目前文献可得,至少已鉴定出52种不同的SLC9A6致病变异,其中包括4种X染色体片段缺失变异和10种内含子剪接位点变异,错义变异26例,无义变异12例。外显子突变谱显示12号外显子是致病变异的高发区,占所有外显子致病变异的43.8%。NHE6是一种十二个跨膜结构域组成的跨膜蛋白,12号外显子编码最后一个跨膜基序,突变谱表明12号外显子在SLC9A6的功能中起着至关重要的作用[12]。本文所报道病例基因突变是位于11号内含子的经典剪切位点,既往尚未报道,可推测由于剪切突变导致SLC9A6基因剪切异常,影响12号外显子,从而导致疾病发生。错译变异、无义变异对内体和细胞功能的影响较小,可能不会导致蛋白质表达或功能的完全丧失,但可产生功能特性改变(显性抑制或组成性激活)的蛋白产物,有文章推测这些产物可与其他遗传因素一起,导致Christianson综合征患者疾病严重程度的变化[13]。2020年Ilie等[13]描述了2种SLC6A9错义变异或无义变异如何损害NHE6活性和内体功能,表明此类突变体在生物合成翻译后成熟、膜分选、再循环内体中的pH稳态和蛋白运输方面表现出不同的缺陷,并且还可引发细胞凋亡。
Na+/H+交换蛋白(Na+/H+ exchanger,NHE)家族由九个成员组成,NHE1-5在细胞质膜,NHE6-9在细胞内的细胞器膜中。SLC9A6所编码的NHE6位于早期再循环的内体膜中,但并不存在于溶酶体中,可在细胞膜表面有短暂的低表达[14]。有研究表明NHE异构体的过表达增加了此类蛋白质所在腔室的腔内pH值,NHE6在早期的再循环内体膜中可调节pH值,使其内腔由微酸环境变为胞质中性环境,因此,NHE6失活的一个可能的结果是降低再循环内体腔的pH值,另一方面也降低了腔内单价离子的含量,这两者都可能影响蛋白质折叠和运输[14,15]。近年的研究表明,NHE6蛋白基因敲除的神经元中显示细胞内定位于胞体的溶酶体数量减少以及溶酶体蛋白酶活性的降低。自噬流分析显示,与野生型相比NHE6蛋白基因敲除的神经元的自噬流减少了50%以上;NHE6缺失大鼠脑细胞显示自噬体的异常积聚[16,17,18]。再循环内体运输对于长时程增强(long-term potentiation,LTP)过程中树突棘的生长是必不可少的,这个过程被认为是学习和记忆的分子基础[19]。在NHE6蛋白基因敲除的小鼠尸检中,星形胶质细胞和小胶质细胞表现出明显的增生和神经胶质反应,且主要集中在轴突中[20],这表明SLC9A6对于维持神经元极性和成熟的突触结构至关重要。2021年Lizarraga等[21]发现,无论突变如何,所有患者来源的神经元都表现出轴突生长和分支形成减少,这证明了上述观点的同时,也表明其可能是患者出现小头畸形的原因。NHE6调节细胞活力的机制虽尚未阐明,然而从最近的研究中我们得知,NHE6和神经营养因子TrkB受体共定位于小鼠海马神经元的再循环内体中[13]。Trk家族的神经营养因子受体是神经系统发育、存活和可塑性的正向调节因子[22]。因此,如果SLC9A6的突变损害了内体运输,会导致再循环内体的过度酸化和TrkB受体信号传导、神经元树突分支、突触数量和连接减少。此类病理改变可能是导致Christianson综合征患者发育迟缓和倒退、智力残疾、癫痫、共济失调、小脑和脑干萎缩等一系列神经、躯体和行为症状的机制。2019年Pescosolido等[23]报道SLC9A6变异的女性携带者已被诊断出通常与tau蛋白沉积相关的神经变性有关,NHE6活性丧失可能有助于了解Tau蛋白病的分子基础和核内体在神经变性中的作用,其致病机制有待进一步研究。
到目前为止,Christianson综合征尚无被证实有效的治疗方法,近年有文献提出:(1)可以根据癫痫的发作形式选择相应的抗癫痫发作药物,并根据治疗效果进行调整,药物难治性癫痫可选择生酮饮食治疗;针对精神运动发育迟滞,可以进行康复训练。(2)将来对CS有两个治疗方向,一是基因治疗,二是根据Christianson综合征的发病机制进行相应的处理,譬如有学者提出,NHE6蛋白缺失导致TrkB信号衰减造成的相关功能缺陷可以通过高水平外源性TrkB来激活TrkB受体进行挽救[24]。本例患儿以癫痫和全面发育迟缓为核心症状,给予抗癫痫发作药物口服、康复训练等,癫痫发作控制,发育缓慢进步,但仍有全面发育迟缓。国内尚无针对该病的精准治疗手段。从目前报道的病例来看,其临床结局与表型之间有一定联系,存在发育倒退、难以控制的频繁癫痫发作以及合并消化、呼吸等其他系统异常的患者预期寿命有限,但仍需进一步研究。
综上所述,本文报道了1例诊断为Christianson综合征的患儿,增加了患者的临床表型,对有癫痫合并全面发育迟缓、发育倒退的患儿,需考虑该疾病,及时进行遗传学检测明确先证者。若家庭有再生计划,强调产前诊断的重要性。本例通过WES技术发现了一个新的X染色体长臂SLC9A6基因c.1366+1G>C剪切变异,扩展了该病基因变异谱,对于本病的发病机制期待进一步深入研究。
所有作者均声明本研究不存在利益冲突
董燕,连若斐,金亮,等.SLC9A6基因突变致Christianson综合征患儿临床及遗传学分析[DB/OL].中国临床案例成果数据库,2023(2023-02-22).http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2023.e00719.

























