
先证者,女,1岁1个月,因"发热、呕吐、发现低血糖6 d"就诊。
患儿入院前6 d发热伴呕吐,抽搐1次,抽搐后嗜睡,测血糖低。静脉应用葡萄糖后症状缓解。体格检查:神志清,反应欠佳,心腹及神经系统体格检查未见明显异常。病程中间断发热并再次发生低血糖时未出现抽搐或睡眠异常等现象。既往体健。
住院期间血尿遗传代谢筛查提示游离肉碱缺乏,完善全外显子基因检测:ACADM基因存在两个杂合变异:c.449_452delCTGA(p.T150Rfs*4)和c.1085G>A(p.G362E)。经PCR-Sanger测序方法验证,这两个突变分别来自患儿父母,且为致病性变异。1周后复查血遗传代谢筛查示己酰肉碱(C6),辛酰肉碱(C8)、C8/C10均显著增高。故诊断为中链酰基辅酶A脱氢酶缺乏症。
口服左卡尼汀,避免长时间禁食、避免感染。
无低血糖发作,无抽搐,生长发育正常。
儿科;内分泌科;重症医学科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
中链酰基辅酶A脱氢酶缺乏症(medium-chain acyl-coenzyme A dehydrogenase deficiency,MCADD)由ACADM基因(OMIM 607008)纯合或杂合突变引起的脂肪酸氧化缺陷,是一种常染色体隐性遗传疾病[1]。急性发作常表现为呕吐、低酮症性低血糖,也可表现为高氨血症、嗜睡、昏迷、癫痫发作甚至猝死等。该病临床表现异质性大,早期诊断困难,容易误诊,死亡率高。本文对我院诊治的1例MCADD患儿的临床特征和基因进行分析,总结相关文献,以提高医务工作者对MCADD的早期识别。
患儿,女,1岁1个月,因"发热、呕吐、发现血糖低6 d"于2018年7月18日入院。入院前6 d患儿无明显诱因出现发热,热峰39.4℃,伴呕吐,抽搐1次,表现为双眼上翻凝视,口周发绀,四肢强直抖动,持续约1 min后自行缓解,缓解后患儿嗜睡,无大汗、皮肤湿冷等现象。时测血糖示1.3 mmol/L,给予静脉注射10%葡萄糖注射液后患儿觉醒,意识恢复正常,复测血糖为12 mmol/L。在当地诊断"中枢神经系统感染、低血糖"收入院。在当地住院5 d,期间监测血糖又发现1次低血糖,空腹血糖1.8 mmol/L,患儿无昏迷或抽搐,无异常哭闹,予以进食后复测血糖达5.6 mmol/L,后监测血糖均正常。患儿未再呕吐,精神尚可,仍发热,转诊来我院。病后大小便外观、次数均无异常。
体格检查:体重11 kg,身高76 cm,神志清,精神欠佳,全身皮肤温暖,未见皮疹,头颅无畸形,头围46 cm。心肺腹及神经系统体格检查未见异常。
既往无特殊病史,第1胎第1产,足月顺产,出生体重3.2 kg。生长发育正常,无抽搐、昏迷、血糖异常等家族史。
血常规、肝肾功能、心肌酶未见异常。血糖监测未见异常,脑脊液常规、生化未见异常。血气分析:pH 7.378,PO2 103 mmHg(1 mmHg=0.133 kPa),PCO2 28.6 mmHg,BE -7.3,乳酸2.0 mmol/L。血氨8.7 mmol/L。尿酮体阴性。尿有机酸分析:未见典型有机酸尿症改变。3-羟基丁酸、乙酰乙酸、辛酸、3-甲基戊烯二酸,3-羟基戊二酸、乙酰甘氨酸、辛二酸浓度稍高,可能与食物药物代谢相关。血游离肉碱及酰基肉碱筛查:入院时查游离肉碱及多种酰基肉碱浓度显著降低;补充左卡尼汀后复查结果C6、C8、C8/C10均显著增高,符合MCADD(表1)。腹部及心脏超声未见异常。心电图、脑电图、头颅磁共振平扫未见异常。
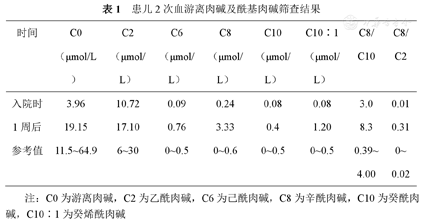
患儿2次血游离肉碱及酰基肉碱筛查结果
患儿2次血游离肉碱及酰基肉碱筛查结果
| 时间 | C0(μmol/L) | C2(μmol/L) | C6(μmol/L) | C8(μmol/L) | C10(μmol/L) | C10∶1(μmol/L) | C8/C10 | C8/C2 |
|---|---|---|---|---|---|---|---|---|
| 入院时 | 3.96 | 10.72 | 0.09 | 0.24 | 0.08 | 0.08 | 3.0 | 0.01 |
| 1周后 | 19.15 | 17.10 | 0.76 | 3.33 | 0.4 | 1.20 | 8.3 | 0.31 |
| 参考值 | 11.5~64.9 | 6~30 | 0~0.5 | 0~0.6 | 0~0.5 | 0~0.5 | 0.39~4.00 | 0~0.02 |
注:C0为游离肉碱,C2为乙酰肉碱,C6为己酰肉碱,C8为辛酰肉碱,C10为癸酰肉碱,C10∶1为癸烯酰肉碱
基因检测:经患儿父母知情同意后分别抽取患儿及其父母外周静脉血3ml,以EDTA抗凝,行全外显子组的基因测序及Sanger测序。全外显子基因分析发现ACADM基因存在两处杂合变异:c.449_452delCTGA(缺失突变)和p.T150Rfs*4(移码突变)。这两个变异均已有相关的文献报道[2,3]。c.449_452delCTGA(p.T150Rfs*4)突变导致蛋白翻译提前终止,对蛋白功能的影响可能较大;c.1085G>A(p.G362E)在健康人群携带率极低。根据美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)发布的变异解读指南进行致病性分析,两个变异均为致病变异。家系验证结果显示这两个变异分别来自于父母,为复合杂合变异,符合常染色体隐性遗传(图1)。
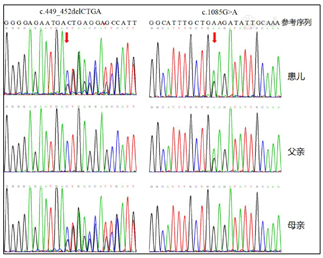
中链酰基辅酶A脱氢酶缺乏症:本例患儿发热后出现呕吐、抽搐、嗜睡,低酮症性低血糖发作,血游离肉碱及酰基肉碱复查结果为C6、C8及C8/C10升高,基因检测提示ACADM基因存在杂合变异,为致病性变异,故诊断成立。
(1)戊二酸血症Ⅰ型:又称戊二酸尿症Ⅰ型,是由于细胞内戊二酰辅酶A脱氢酶缺乏导致戊二酸蓄积,引起代谢紊乱及神经系统损害。本病常表现为感染、高蛋白饮食、疲劳或预防接种后加重,出现酮症、呕吐、昏迷或惊厥、肝大等表现。急性期可出有代谢性酸中毒、低血糖、酮症、高氨血症,血质谱分析结果提示戊二酰肉碱增高,游离肉碱降低,可通过基因检测明确。本患儿呕吐、低血糖发作,伴有抽搐、嗜睡,查游离肉碱下降,故需鉴别,但患儿查戊二酰肉碱未见增高,而且基因检测结果不支持本病。
(2)原发肉碱缺乏症:称原发性肉碱吸收障碍或肉碱转运障碍,是由于SLC22A5基因突变引起。可有低血糖发作、抽搐、心肌病、肌无力等临床表现。实验室检查代谢性酸中毒、转氨酶及肌酸激酶升高、高血氨、血糖低等,血游离肉碱及酰基肉碱检测会出现游离肉碱明显下降,通过基因检测可确诊。本患儿有呕吐、低血糖发作,伴有抽搐、嗜睡,查血游离肉碱及酰基肉碱第一次结果示血游离肉碱明显下降,故需鉴别,但复查血质谱分析结果提示C6、C8、C10均升高,而且基因检测结果不支持本病。
(3)Reye综合征:以急性非炎症性脑病伴高氨血症、肝功能障碍和肝脏脂肪浸润为特征。多与感染、中毒、遗传代谢异常及药物应用尤其是水杨酸类有关。临床表现为意识障碍、惊厥、肝功能异常、低血糖。本例患儿有呕吐、意识丧失发作,低血糖需鉴别,但患儿无阿司匹林应用史,意识丧失经过静脉补充葡萄糖后很快恢复。查肝功未见异常,血氨无升高,脑电图及头颅磁共振未见异常,不支持诊断。
左卡尼汀口服液100 mg/(kg·d),分3次口服。1周后左卡尼汀改为50 mg/(kg·d)口服。出院后叮嘱家长避免患儿长时间禁食,加强护理,避免感染。
出院后每日2次监测血糖,后改为每周1次监测血糖均正常。每3个月复查1次血游离肉碱及酰基肉碱,指导调整左卡尼汀用量。定期遗传代谢病专科复诊。现随访1年患儿无低血糖、抽搐发作,智力、运动发育正常。
儿童低血糖是临床常见的代谢紊乱,发生率约为10%,其中遗传代谢性疾病相关低血糖的发病率为1/30 000~10/30 000[4]。引起低血糖常见的遗传代谢病为高胰岛素相关代谢异常、脂肪酸氧化障碍、氨基酸代谢异常、糖原代谢异常、葡萄糖代谢异常等。脂肪酸氧化障碍主要包括肉碱转运酶缺陷和遗传性生酮缺陷[4]。遗传性生酮缺陷主要分为三大类:(1)短链酰基辅酶A脱氢酶缺乏症;(2)中链酰基辅酶A脱氢酶缺乏症;(3)长链(极长链)酰基辅酶A脱氢酶缺乏症。
MCADD是由基因ACADM编码,位于染色体lp31。ACADM基因的突变存在种族差异,在白种人患者中,c.985A>G突变最常见,占80%~90%[5,6]。c.449_452delCTGA是亚洲人ACADM基因中最常见的突变位点[7,8,9]。当两个等位基因均发生突变时,所编码的中链酰基辅酶A脱氢酶合成减少甚至缺乏,导致中链脂肪酸氧化代谢障碍。这时MCADD患者需通过消耗糖原储备来补偿机体能量供给,然而在禁食或疾病期间,糖原储备会迅速耗尽,而且脂肪酸氧化缺陷、乙酰辅酶A产生减少会限制糖异生过程,进而引起低血糖加剧和失代偿[6,10,11]。MCADD的发病率因地域不同而有差异。MCADD更常见于高加索人群,发病率为1∶10 000~1∶27 000[12,13]。美国该病的发病率为1∶10 000~1∶30 000[14],德国的发病率为1∶9773[15]。亚洲发病率较低,其中日本约为1∶108 000[16],中国台湾地区新生儿筛查中患病率为1∶263 500[17]。中国南方人群新生儿筛查发现MCADD患病率为1∶222 902[18]。
MCADD是否发病受很多因素影响比如饥饿、感染、免疫接种等。本病首次发作多在4个月~4岁之间,有些MCADD患者可能整个童年时期不会发病,也有些患者首次发作时病情很严重,约24%的患者首次发作即出现死亡[19]。本病临床表现与基因突变型有一定关系,研究发现c.985A>G纯合突变患者血浆辛酰肉碱水平较高,更容易发生不良事件,ACADM中基因突变的其他类型与临床表现之间的关系尚未得到明确[20,21,22]。出现临床症状的MCADD患者一般都有诱发因素,长时间禁食是该病最常见的诱因。急性发作时,约有90%的患者出现低血糖,有些可出现严重低血糖,约一半患者出现呕吐和肌无力,也可出现其他症状高氨血症、嗜睡、昏迷、癫痫发作甚至猝死[23]。需要注意与Reye综合征鉴别。本例患儿生后无临床症状,于1岁1个月时发病,因感染诱发了低血糖发作、抽搐、呕吐。
目前国内外推荐使用干血斑串联质谱法检测C6(己酰肉碱)、C8(辛酰肉碱)、C10(癸酰肉碱)及C8/C10来诊断本病,其中血C8升高是MCADD生化诊断的主要标志,C6、C10、C8/C2、C8/C10升高是MCADD生化诊断的典型标志[19,24,25]。但脂肪酸代谢性疾病可消耗肉碱,引起继发性肉碱缺乏,导致血清C0降低,甚至合并多种酰基肉碱降低,这时C6、C8、C10可能不会升高,可以通过酰基肉碱之间的比值或结合ACADM基因检测进行鉴别[26]。本例患儿病初游离肉碱及酰基肉碱检测未见C6、C8、C10升高,也未见C8/C2、C8/C10比值的升高,考虑与肉碱过度消耗有关。Gong等[27]总结了24例MCADD患儿的特点,其中有1例患儿为7月龄,血斑质谱分析示C0显著降低(C0:2.72 μmol/L),C6、C8、C10及C8/C10均未见明显升高,后经基因证实为本病,与本患儿类似。本例患儿在予以左卡尼汀补充1周后复查血游离肉碱及酰基肉碱筛查提示C0恢复正常,此时C6、C8及C8/C10均明显高于正常值,因此在临床中遇到游离肉碱显著减低的患儿,需在应用左卡尼汀后及时复查血酰基肉碱谱并结合基因检测进行明确诊断。取成纤维细胞或其他组织(肝脏、心脏、骨骼肌)检测MCAD的活性减低(<10%)是确诊MCADD的"金标准"[28]。但因其为有创检测,限制了在临床中的应用。因此临床常借助ACADM基因测序诊断本病。本例患儿基因测序发现ACADM基因存在两处杂合变异,明确诊断为MCADD。
MCADD是可治疗的遗传代谢病,如能早期诊断,预后较好。主要治疗原则为避免长时间禁食,急性发作期积极对症处理。建议最长禁食时间因年龄而异,6~12个月不超过8 h,1~2岁不超过10 h,2岁后不超过12 h[29]。反复出现低血糖的幼儿可在夜间给予补充复杂的碳水化合物(例如生玉米淀粉,2 g/kg)。目前仅建议对血清肉碱水平低的MCADD患者进行肉碱补充[6]。本例患儿病初因肉碱缺乏,故予以补充,后复查正常后停用。MCADD存在猝死风险,当患者疾病状态或长时间禁食时,应立即给予补充葡萄糖,如出现低血糖症状更要及时补充。MCADD患者缺乏能够代谢中链脂肪酸的酶,理论上应减少脂肪摄入,但仍存争议,因为没有证据表明膳食中的中链脂肪酸会对MCADD患者造成伤害[24]。研究表明,MCADD患者引发肥胖的风险较高,建议健康饮食,多运动[29]。
综上,MCADD发病率地域间差别较大,亚洲人发病较少。长时间禁食、感染等可诱发MCADD患儿出现嗜睡、抽搐、低血糖发作表现,血游离肉碱及酰基肉碱检测可辅助诊断,结合ACADM基因测序可明确诊断,早期诊断、及时干预可显著改善预后。
王檬,王海军,李东晓,等.中链酰基辅酶A脱氢酶缺乏症1例[DB/OL].中国临床案例成果数据库,2023(2023-03-14).http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2023.e00936.
所有作者均声明本研究不存在利益冲突

























