
患者,男性,33岁,因发现双侧睾丸大小不一致18个月伴体重增加20斤入院。
专科检查发现男性乳房发育伴血清雌二醇水平升高及睾酮水平降低,射精无精子。
门诊超声提示:双侧睾丸实性占位性病变伴钙化。核磁共振显示双侧睾丸大小及信号异常。行双侧睾丸切除术后送入病理科。大体见双侧睾丸肿块,切面灰白间黄,左侧伴囊性变。镜下双侧睾丸病变形态基本一致,肿瘤呈结节状生长,排列呈巢片状、条索状,瘤细胞主呈上皮样,胞质丰富、嗜酸性或淡染,细胞核大,核仁明显,间质黏液变性及玻璃样变伴部分区域可见钙化。免疫组化染色显示肿瘤细胞表达S100,α-Inhibin及Calretinin。下一代测序技术检测肿瘤细胞存在PRKAR1A基因体细胞突变,未检出胚系突变。最终病理诊断为双侧睾丸大细胞钙化性支持细胞瘤。
行双侧睾丸及肿瘤切除术。
术后2周随访患者,健康状况良好,无肿瘤复发或转移。
病理科;泌尿外科;肿瘤科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
大细胞钙化支持细胞瘤(Large-cell calcifying Sertoli cell tumors,LCCSCT)是一种罕见的睾丸性索间质肿瘤,主要发生于年轻患者。自1980年首次描述以来,目前报道的病例不足100例。临床多为散发,部分病例与内分泌综合征相关,最常见伴Carney综合征和Peutz-Jeghers综合征,约40%的病例累及双侧睾丸。临床上表现为睾丸肿大,性早熟以及乳房发育,性功能低下以及内分泌综合征的症状。生物学行为多为良性,恶性行为少见[1,2]。由于LCCSCT罕见,临床及病理医师对其缺乏认识,诊断困难,且部分病例和遗传综合征相关,准确的诊断对于治疗及遗传学监测非常重要。我们报道1例双侧睾丸的LCCSCT,结合相关文献对其临床病理特点分析及探讨,旨在提高对临床及病理医生对本病的认识。
患者,男性,33岁,因发现双侧睾丸大小不一致1年半入院,体重近1年增加20斤。门诊查超声提示:左侧睾丸35.8 mm×17.9 mm,内见一大小约18 mm×16 mm低回声团,右侧睾丸50.7 mm×42.9 mm,内部见小范围不规则无回声区及多个点状强回声,考虑实性占位性病变伴钙化。核磁共振T2信号显示双侧睾丸大小及信号异常(图1A)。专科检查:阴茎阴囊发育可,双侧阴囊内可触及睾丸,左侧约3 cm,右侧约5 cm,未触及明显肿块,触之不痛;男性乳房发育,血清雌二醇升高(100.40 pg/ml);泌乳素升高(14.26 ng/ml),睾酮降低(0.99 ng/ml)。射精无精子。既往有肾结石手术史,无家族病史和肿瘤病史。
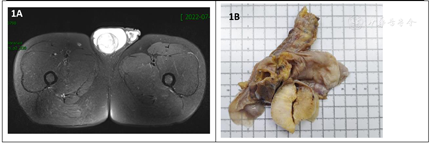
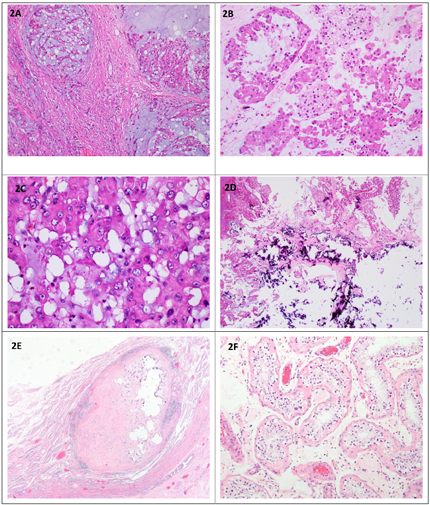
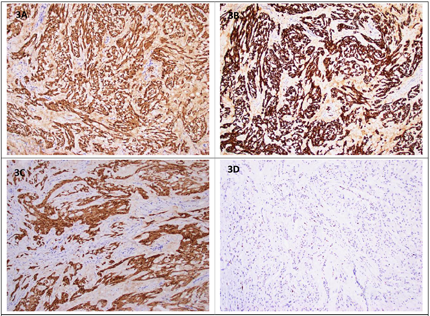
患者入院后行双侧睾丸病变切除术,术后送病理检查。大体所见:右侧睾丸,大小5 cm×4.5 cm×4.5 cm,表面光滑,临床已剖开,包膜完整,切面灰白间黄,几乎均为肿块(图1B),相连附睾4.5 cm×1 cm,切面暗红,充血,相连精索长7 cm,直径1 cm;左侧睾丸,大小3 cm×2.5 cm×2 cm一个,切开见2 cm×1.5 cm囊腔,内容凝血块,相连附睾3.5 cm×1.2 cm,切面暗红,充血,相连精索长7 cm,直径1cm,另见游离灰白肿块样物2.5 cm×1.5 cm×1 cm。经取材制片后显微镜下观察。镜下见双侧睾丸病变基本一致,肿瘤呈多结节状生长(图2A),细胞排列呈巢片状或条索状,细胞主呈上皮样,胞质丰富、嗜酸性,部分胞浆淡染、空泡状(图2B),细胞核大,核仁明显(图2C),间质伴黏液变性及玻璃样变,部分区域可见钙化(图2D);右侧睾丸肿瘤周边组织内可见肿瘤性小结节(图2E),周边睾丸组织中生精小管未见生精现象(图2F)。左侧睾丸肿瘤间质玻璃样变明显,临近肿瘤细胞间质可见黏液变性伴中性粒细胞浸润。基于以上形态,结合患者年龄、肿瘤发生部位及形态学特征,我们考虑了以下肿瘤:1.性索-间质细胞肿瘤;2.肾上腺残余瘤;3.生殖细胞肿瘤;4.上皮源性肿瘤;5.其他;按照初步诊断思路,行免疫组化检测PCK、S-100、CD99、α-Inhibin、Calretinin、MelanA、AR、SALL4、Ki67,结果显示S-100阳性(图3A),a-Inhibin(图3B)及Calretinin阳性(图3C),MelanA、SALL4、CD99、AR及PCK均阴性,Ki67增值指数约1%(图3D),符合睾丸性索-间质细胞肿瘤的免疫表型。
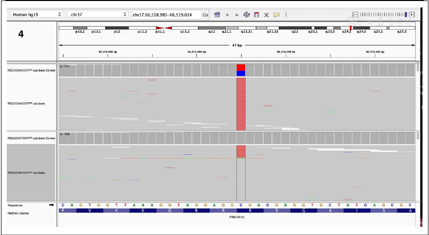
下一代测序(Next Generation Sequencing,NGS)技术检出肿瘤细胞PRKAR1A基因的第96位氨基酸由精氨酸突变成终止子(C.286C>T, p.Arg96*)。同时未检出胚系突变(图4)。
该例结合患者临床情况、经典的病理形态学及免疫组化检测结果,符合睾丸大细胞钙化性Sertoli细胞瘤。鉴别诊断包括:1.睾丸间质细胞瘤(Leydig细胞瘤)睾丸Leydig细胞瘤肿瘤排列呈巢状、片状、条索状,胞质嗜酸性,约40%的病例可见Reinke结晶,肿瘤细胞内可见脂褐素,免疫组化a-Inhibin、Calretinin、MelanA及CD99阳性,S100通常阴性。本例虽临床表现与Leydig细胞瘤有重叠,但双侧睾丸起病,形态学有明显黏液样间质及钙化,且无Reinke结晶及细胞内脂褐素,免疫组化Melan-A及CD99阴性,S100阳性,不支持睾丸Leydig细胞瘤[2,3,4]。2.睾丸其他类型支持细胞瘤(Sertoli细胞瘤)依据2016版泌尿及男生殖系统WHO,睾丸Sertoli细胞瘤包括三种类型:Sertoli细胞瘤,非特指型、LCCSCT和生精小管内大细胞玻璃样变的Sertoli细胞瘤,硬化性Sertoli细胞瘤已归入非特指型[4]。非特指类型支持细胞瘤大多发生在中年人,肿瘤呈多结节状,形态多样,从分化良好的圆形中空管腔至发育不良的实性小管,总能见到特征性小管结构,小管周围基底膜物质围绕;肿瘤细胞胞质中等至丰富,透明或嗜酸性,偶尔富含脂质而呈空泡状,核糖圆形或长形,核沟及核内包涵体不常见,核仁不明显,这与本例形态有所不同。非特指型Sertoli细胞瘤及睾丸生精小管内大细胞玻璃样变的Sertoli细胞瘤罕见有钙化灶出现,而在LCCSCT中多发性钙化是一个可靠而重要的特征性诊断依据[5]。
患者于2022年8月入院后,综合多方面因素考虑,遂行双侧睾丸及肿瘤切除术。
患者手术后处于恢复期,目前生活质量良好。因随访时间较短,有待于进一步随访观察。
LCCSCT是睾丸罕见的性索间质肿瘤,1980年由Proppe等学者首次报道[6],迄今全球报道不足100例,多为个案和小宗案例报道[1,2,6]。LCCSCT平均发病年龄约16岁,主要体征是睾丸的结节状肿块,部分病例两侧睾丸均有病变。一些患者同时患有遗传综合征,伴有异常性早熟、乳房发育及性功能低下等内分泌改变[1,2,3,4,5,6,7]。LCCSCT伴发的临床综合征通常是Peutz-Jeghers综合征和Carney综合征,Peutz-Jeghers综合征又叫黑斑息肉病,半数病例有家族遗传史,常以LKB1/STK11基因突变为特征,表现为消化道的非肿瘤性息肉和皮肤斑点。睾丸生精小管内大细胞透明变支持细胞瘤被认为是一种与Peutz-Jeghers综合征相关的独特实体,具有STK11基因的特征性突变。Carney综合征是显性遗传病,常见PRKA1A基因的失活突变,表现为斑点性皮肤色素沉着、内分泌病和多种肿瘤,包括心脏黏液瘤、垂体腺瘤、甲状腺癌和神经鞘瘤等[1]。本例患者在右侧睾丸肿瘤旁局部查见生精小管内大细胞透明变支持细胞瘤样病变,提示可能与Peutz-Jeghers综合征相关,但该患者并无Peutz-Jeghers综合征与Carney综合征相关症状的体征及家族史,基因检测发现肿瘤细胞出现PRKA1A基因无义突变,但未检出胚系突变。进一步证明该病例为散发病例。这一点对随访工作有重要的指导意义。
多数LCCSCT属于良性肿瘤,少部分LCCSCT生物学行为恶性,恶性常发于成年人,表现为单侧、孤立肿物。2016版WHO男性生殖与泌尿肿瘤分类中指出:当肿瘤出现以下形态学特征中两项及两项以上时提示具有恶性生物学行为:①肿瘤最大径>4cm;②显著核异性;③核分裂增多(>3个/10HPF);④肿瘤性坏死;⑤出现淋巴管血管侵犯;⑥睾丸外侵犯。本病例肿瘤最大径5cm,检出脉管内瘤栓,提示肿瘤可能具有恶性生物学行为,建议临床密切随访[4]。
LCCSC发生于双侧睾丸和多灶性病变时常提示为与遗传综合征相关。镜下形态学表现为肿瘤细胞呈巢状、条索状和实性小管状分布,伴黏液样或胶原性的间质,肿瘤细胞胞质丰富嗜酸性,部分瘤细胞的胞质中可以见到一些小空泡,细胞核圆形,核大空泡状,核仁明显,除恶性病例外,核分裂象少见。瘤细胞巢由含有大片钙化的丰富纤维间质所分隔,钙化是本瘤的一个重要特征。LCCSC免疫组化表达S100,α-Inhibin及Calretinin[5]。本例具有经典的LCCSC形态学与免疫表型,但未检出相关基因的胚系突变。
有研究表明,免疫标记物β-Catenin在非特指类型的支持细胞瘤呈核阳性,而在LCCSCT及管内透明变性支持细胞瘤内呈核阴性,有助于支持细胞瘤亚型之间的鉴别诊断[8]。一项最新的研究通过对15例LCCSCT病例中PRKAR1A免疫组化表达研究,证明了PRKAR1A可能是LCCSCT的一种敏感和特异性的生物标志物,PRKAR1A缺失可能有助于其诊断,特别是在散发性病例和那些是Carney综合征病例中,但其敏感性和特异性在病理诊断中的应用还有待于更多数据的积累[10]。
LCCSCT目前主要采用手术切除睾丸及肿瘤,良性肿瘤预后好。虽然睾丸肿瘤行睾丸切除是标准的治疗术式,但由于生理的特殊性,LCCSCT行部分切除睾丸肿瘤切除术似乎是对患者更有益的手术方法,本例患者已生育一子,本人无再生育意愿,且精液检查提示为无精症,要求行双侧睾丸切除术,综合其生育需求及治疗意愿,遂行双侧睾丸切除。
本病例肿瘤周边睾丸组织生精小管内未见生精现象,可能由于肿瘤压迫及肿瘤细胞分泌芳香化酶使雄激素转化为雌激素(患者雌激素升高),进而导致生精减少或生精停滞以及乳腺发育。文献报道,通过降低雌激素水平治疗乳腺癌,可用于治疗LCCSCT相关的乳腺发育[11]。有研究表明,阿那曲唑是一种芳香化酶抑制剂,芳香化酶抑制剂可能通过增强芳香化成为患者的有效治疗模式,但目前只有有限数量的患者接受了治疗,尚需要积累临床试验的数据进一步验证[1]。
本文总结了1例年轻患者睾丸LCCSCT临床病理特点并进行文献回顾,但由于笔者在整理病例时,该患者术后两月,尚处于恢复期,身体状况良好,尚缺乏长期随访观察数据,在未来的工作中还需跟踪观察。此外,本例患者下一代测序技术检出肿瘤细胞PRKAR1A基因突变,但未检出相关基因的胚系突变,提示该病例为散发病例。这也是该病例的特殊之处。同时,分子遗传学检测结果为患者后续治疗及随访管理提供重要的参考依据。
陈烁,陈伟斌,尚士宣,等.双侧睾丸大细胞钙化性Sertoli细胞瘤1例[DB/OL].中国临床案例成果数据库,2023(2023-03-17).http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2023.e00955.
感谢美国Tufts大学医学院病理系主任周敏教授对该病例病理诊断的指导
所有作者均声明本研究不存在利益冲突

























