
患儿系4+月龄男性婴儿,因"咳嗽20 d,加重伴发热10 d"就诊。
急性重病容,T 38.3℃,P 182次/min,R 78次/min,BP 89/43 mmHg(1 mmHg=0.133 kPa),格拉斯评分13分,面罩吸氧下SPO2 86%,肛周可见散在皮疹,全身浅表淋巴结未扪及肿大,结膜正常,瞳孔等大等圆,对光反射正常,鼻翼扇动,口唇发绀,咽部充血。呼吸运动对称,吸气性三凹征阳性,呼吸音粗糙,双肺可闻及喘鸣音及少量湿啰音。全腹柔软,肝脏肋下2 cm触及,缘钝质软,脾脏未触及,神经系统查体无异常。
患儿入院后查呼吸道合胞病毒阳性,胸部CT示:双肺感染,细胞免疫:CD3 2%~3.3%,CD4 0.9%~1.8%,CD8 0.3%~0.4%,CD4/CD8 2.3~6,B淋巴细胞95.5%~95.9%,NK细胞0.6%~0.7%。体液免疫IgA<0.07 g/L。家系全外显子测序分析查见IL2RG基因c.1A>T(母源)杂合变异,致病性评级为可能致病,故诊断X-连锁重症联合免疫缺陷病。
患儿入院后立即予以严密监护及对症治疗,同时先后予泰能、亚胺培南西司他丁、万古霉素抗细菌感染,米开民、伏立康唑预防真菌感染,磺胺预防卡氏肺囊虫肺炎感染,并输注丙种球蛋白,效果欠佳。根据其基因检测结果,建议行造血干细胞移植治疗。
患儿抗感染治疗效果欠佳,基因检测结果回示后,家属拒绝行造血干细胞移植,要求出院,不久后患儿死亡。患儿父母再孕二胎行羊水产前诊断,提示胎儿性别为男性,Sanger测序未查见胎儿羊水细胞IL2RG基因c.1A>T突变,遂妊娠至足月分娩。现二孩3+月龄,体健。
血液科;儿科;遗传科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
X-连锁重症联合免疫缺陷病(X-linked severe combined immunodeficiency disease,X-SCID是原发性免疫缺陷的一种,其特征是T和B淋巴细胞系统的严重缺陷。由Noguchi等[1]于1993年首次报道,目前统计发病率约为1/50000-1/100000[2],常见于男性,是一种罕见的致死性疾病。患者常见临床表现为免疫系统缺陷导致的各种机会性感染,普通抗感染治疗效果欠佳,经造血干细胞移植或基因治疗可获得缓解。本文报道了一例新发现的由IL2RG基因c.1A>G(母源)突变导致的以肺炎为首发表现的X-连锁重症联合免疫缺陷病患儿,明确诊断后对其母亲再孕二胎进行了产前诊断。旨在提高对本病的认识,以期尽早诊断、治疗,改善患者预后及降低家庭再生育风险。
患者系4+月龄男性婴儿,因"咳嗽20 d,加重伴发热10 d"就诊。20 d前患儿无明显诱因出现轻微咳嗽,可闻及喉间痰响,伴流涕,无发热、气促、奶量下降、呕吐、腹泻、神萎、抽搐等症状。后患儿咳嗽症状逐渐加重,并出现奶量下降,情绪烦躁不易安抚,外院就诊胸片提示:双肺纹理稍多,未见实质性浸润灶,两次痰培养阴性。予阿莫西林克拉维酸钾及头孢哌酮他咗巴坦抗感染及雾化对症治疗,未见明显好转。10 d前患儿出现反复发热(最高体温39.6℃),咳嗽流涕症状进一步加重,伴气促、喘息、精神差,无呕吐、腹泻、抽搐等。1d前患儿出现心率、呼吸明显增快,体格检查:T 38.3℃,P 182次/min,R 78次/min,BP 89/43 mmHg(1 mmHg=0.133 kPa)。入院可见急性重病容,神志清楚,面罩吸氧下SPO2 86%,肛周可见散在皮疹,无皮下出血、水肿,全身浅表淋巴结未扪及肿大,结膜正常,瞳孔等大等圆,对光反射正常,鼻翼扇动,口唇发绀,口腔黏膜正常,咽部充血。颈软,呼吸运动对称,吸气性三凹征阳性,呼吸音粗糙,双肺可闻及喘鸣音及少量湿啰音。心律齐,心音有力,未闻及杂音。全腹柔软,肝脏肋下2 cm触及,缘钝质软,脾脏未触及,神经系统查体无异常。既往史、家族史、个人史无特殊。
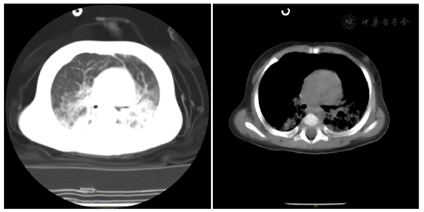
血常规:WBC 13.2×109/L,N 73.7%,HGB 106 g/L,PLT 762×109/L,CRP 9 mg/L。CRP 52 mg/L,PCT 2.13 ng/ml。肝肾功:ALT 47 U/L,AST 58 U/L,TB 0.7 umol/L,ALB 30.5 g/L,LDH 731 U/L,余值大致正常。血气分析:PH 7.340,PCO2 6.0 kPa,K+ 3.22 mmol/L,LAC 2.1 mmol/L,GLU 9.0 mmol/L,余值正常。心肌酶谱未见异常。DIC筛查:DDI 1.22 mg/L,Fg 157 mg/dl,TT 21.5 s,余值正常。胸部CT:双肺感染,有明显渗出和实变及肺间质改变,可见局限性肺气肿;双侧胸膜增厚,右侧胸腔少量积液;双侧肺门显示欠清晰,气管前腔静脉后淋巴结稍显偏大(图1)。多次痰培养:查见革兰阳性球菌、革兰阴性球菌/混合菌丛。呼吸道病毒七联检:呼吸道合胞病毒抗原阳性,其余均阴性。肺炎支原体+衣原体核酸:阴性。体液免疫:IgG 8.5 g/L(输注丙球后),IgA<0.07 g/L,IgM 0.51 g/L,IgE<5 IU/ml。细胞免疫:CD3 2%~3.3%,CD4 0.9%~1.8%,CD8 0.3%~0.4%,CD4/CD8 2.3~6,B淋巴细胞95.5%~95.9%,NK细胞0.6%~0.7%。输血免疫全套:HBsAb、HBeAb、HBcAb阳性,其余均阴性。大便常规未见异常。小便常规:酮体1+,余未见异常。
家系全外显子测序分析查见患儿存在IL2RG基因c.1A>T杂合变异,为与受检者疾病表型相关的可能致病性变异。患儿母亲为该基因变异位点嵌合,二代测序结果提示ref/alt:137/17。患儿父亲、外婆、舅舅及患儿父母再孕二胎经Sanger测序验证均不携带此变异位点(图2)。

患儿为男性,起病急,病程长,临床主要表现为重症肺炎。呼吸道合胞病毒阳性,T淋巴细胞检查CD3、CD4、CD8均显著减低,NK淋巴细胞显著降低,B淋巴细胞虽比例增高,但体液免疫显著减低(IgA<0.07 g/L),回顾胸部CT可见胸腺影小,结合家系全外显子测序分析查见IL2RG基因c.1A>T可能致病性突变,故诊断为X-连锁重症联合免疫缺陷病。
鉴别诊断:1.支气管异物:患儿为小婴儿,有咳嗽、气促、伴喉间痰响,需警惕支气管异物。但患儿系小婴儿,母乳喂养,家属否认有异物吸入史,查体双肺呼吸音对称,胸部CT未见局部性气肿或肺不张征象,故鉴别。2、颅内感染:患儿系小婴儿,以气促、发热、神萎为主要特点,发热时间较长,需警惕,但患儿前囟平,张力不高,无抽搐表现,神经系统查体均阴性,故除外,必要时完善腰椎穿刺以明确。3、其他类型免疫缺陷病:患儿T淋巴细胞显著降低,应与其他影响T细胞功能的原发性免疫缺陷病相鉴别,如X连锁高IgM血症等。患儿IgM无显著升高,且基因检测查见IL2RG基因可能致病性变异,故鉴别。
患儿入院后立即予以心电监护,同时监测24 h出入量,鼻导管吸氧,祛痰、雾化、机械辅助排痰、物理降温等对症治疗,并根据病情进展先后予泰能、亚胺培南西司他丁、万古霉素抗感染,米开民预防真菌感染,丙种球蛋白输注等,效果欠佳。后期患儿有呕吐表现,改米开民为伏立康唑预防真菌感染,磺胺预防卡氏肺囊虫肺炎感染。请血液科会诊考虑:X-连锁重症联合免疫缺陷病可能性大。完善家系全外显子测序分析后查见IL2RG基因c.1A>T(母源)杂合变异,为与受检者疾病表型相关的可能致病性变异。根据基因检测结果,建议行造血干细胞移植治疗。
患儿入院后积极抗感染及对症治疗后症状改善欠佳,体温仍有反复。基因结果回示后患儿家属拒绝行造血干细胞移植,要求自动出院。随访患儿出院后不久即夭折。患儿去世4+年后,患儿父母再次妊娠。通过羊膜腔穿刺术获取羊水细胞行产前诊断,明确受检者父母二胎为男性,Sang测序未查见IL2RG基因c.1A>G突变。现二胎已出生,随访至3+月龄未见明显异常。
重症联合免疫缺陷病(severe combined immunodeficiency disease,SCID)是一组以T细胞发育缺陷导致细胞免疫和体液免疫系统严重受损为特征的罕见遗传异质性疾病[3],新生儿发病率约为1/50000~1/100000[2]。具有发病罕见,预后差等特点。患有SCID的新生儿出生时通常无异常,后期常由于不同类型的感染至医院就诊而得以发现,确诊中位年龄多为4~7月龄[4,5,6]。X-连锁重症联合免疫缺陷病(X-SCID)是最常见的一种SCID,约占50%~60%,除了典型的免疫缺陷感染症状外,主要表现为成熟性T细胞和NK细胞完全性缺乏,B细胞数量则正常或中度增加[7]。
白介素2(interleukin-2,IL-2)细胞因子对维持外周免疫耐受至关重要,其在调节性T细胞(Tregs)发育过程中起重要的作用,同时还正向调节自然杀伤细胞(NK细胞)的细胞杀伤活性以及参与到其他多种细胞因子的信号通路中[8]。白介素-2受体(IL-2R)主要由三部分组成:IL-2Rα、IL-2Rβ、IL-2Rγ,其中IL-2Rγ链在淋巴细胞的增殖和分化中起着重要作用,不仅与IL-2细胞因子相互作用,同时也是IL-4、IL-7、IL-9、IL-15和IL-21细胞因子的共有组成部分。因此,负责编码IL-2Rγ链的IL2RG基因发生突变可导致分化异常的T淋巴细胞、NK细胞以及B细胞功能异常,进而导致X-连锁重症联合免疫缺陷病[1]。
IL2RG基因位于X染色体长臂Xq13区域,共包含8个外显子,编码369个氨基酸(NM_000206.3)。基因突变导致IL-2Rγ链无功能或提前产生终止密码子均可导致淋巴细胞发育过程提前终止。因此在典型的X-SCID患者中会导致多种体液免疫和细胞免疫缺陷,进而使患者极易出现各种细菌或病毒感染,并表现出各种症状如发热、慢性腹泻、皮疹、不能生长发育等等。此外,通常可发现B淋巴细胞功能障碍、低丙种球蛋白血症等[9]。本例报道的患儿以肺部感染为首发症状,具有发热、咳嗽、流涕等常见感染症状,临床症状缺乏特异性,极易与普通婴幼儿肺炎相混淆,后期反复抗感染及对症治疗效果欠佳,症状反复,通过免疫系统检查和基因检测才得以确诊。因此,对于临床上反复感染治疗效果欠佳或怀疑免疫系统缺陷的男性患儿,可尽早行免疫相关检查及基因检测寻找病因。
对IL2RG基因突变导致X-SCID的患者报道最早在1993年[1]。随着测序技术的进步,越来越多的IL2RG基因致病性突变位点被发现。截至目前,HGMD专业版数据库(HGMD Professional 2022.2)已收录310种IL2RG基因导致X-SCID的致病性变异,其中最常见的突变类型为单碱基变异导致的错义或无义突变,共161种。其次为小片段缺失突变,共63种。第三种常见的突变形式为剪接突变,共48种。IL2RG基因编码的8个外显子中每个外显子的致病性变异均有报道,其中以第3、4号外显子变异最为常见。本例患儿为IL2RG基因c.1A>G突变所导致起始密码子变异,影响正常蛋白质编码启动,进而影响基因功能。同时检索对照人群数据库1000Genomes、gnomAD,均未收录此变异。尽管此突变位点此前未有收录,但在同一位点不同碱基变异(c.1A>G)有两例致病性的报道[10,11]。根据ACMG指南,该位点评级为可能致病。同时对家系成员检测发现,受检者母亲存在该杂合变异嵌合,二代测序结果提示ref/alt:137/17。受检者父亲、外婆及舅舅未携带该变异,受检者外公已去世无从验证,整个家系基因检测结果符合X连锁隐性遗传模式。受检者父母再孕二胎行羊水细胞产前诊断明确未携带此变异位点,遂妊娠至足月出生。现已随访至3+月龄,无明显免疫缺陷表现。
X-SCID作为罕见的严重的致死性遗传性疾病,给患者及其家庭带来了巨大的生理、心理压力。目前有效的治疗方法仅有骨髓移植或基因治疗,若不能重建免疫系统,则大多数患儿通常在出生后的两年内死亡[12]。目前国内外已有多例接受造血干细胞移植治疗成功的病例,其中包括非血缘脐血干细胞移植[13]。关于移植治疗时期,有研究提示在患儿3月龄以内进行造血干细胞移植,其五年生存率可达96%[14].因此尽早确诊该类疾病对患儿的预后具有重大意义。本例患儿其父母因社会原因拒绝行造血干细胞移植,自动出院后不久即去世,令人惋惜。如何早期识别并诊断X-SCID,同时建立家属信心,尽早采取造血干细胞移植等手段,是提高患儿生存率的基础。对于已生育X-SCID患儿的家庭,明确基因诊断,并在其再生育时进行恰当的遗传咨询和针对性的产前诊断,是避免再生育患者的重要保障。
本文通过对一例以肺炎为主要表现的患儿的临床表现、免疫检查、基因检测等资料分析,明确诊断了一例X-SCID,提升了对X-SCID疾病的认识。并通过家系全外显子测序分析,发现了一个HGMD数据库此前尚未收录的IL2RG基因c.1A>G突变,丰富了IL2RG基因致病性变异数据库。同时通过产前诊断技术,在患儿父母再孕时明确了二胎未携带此突变位点,缓解了家属的焦虑情绪。在其生产后随访至今,未发现二胎明显异常。该案例期为指导临床医生更好的识别并处理X-SCID疾病,同时对具有相关基因突变的家庭再生育遗传咨询提供依据。
杜雪,徐博成,谢寒冰,等.IL2RG基因突变致X-连锁重症联合免疫缺陷病1例及家系分析[DB/OL].中国临床案例成果数据库,2023(2023-05-11).http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2023.e01380.
所有作者均声明本研究不存在利益冲突

























