
患者,男性,59岁,因"四肢无力半年,构音障碍1个月"入院。患者半年前新冠肺炎疫苗注射后逐渐出现左手无力,肌肉萎缩,并进行性加重;2个月前出现双下肢麻木乏力,伴下肢肌肉萎缩;1个月前出现构音障碍,饮水呛咳,肢体症状较前进一步加重。
舌肌颤动,言语稍含糊
四肢无不自主运动,四肢肌张力减低,肌力:屈颈5级,伸颈5级;肩内收5−级,肩外展5−级;屈腕左3−级,右4−级;伸腕左3−级,右4−级;伸指左2级,右4级;屈指左3级,右4级;握力左3−级,右4级;屈髋5级,伸髋5级;腿内收5级,外展5级;踝跖屈5−级,踝背屈5−级。四肢腱反射活跃,下颌反射(+),左侧Hoffmann征(+),双侧Babinski征(−),双划征(+),脑膜刺激征阴性。闭目难立征(−)。
肌电图示上、下肢及胸旁肌神经源性损害,二代基因测序发现"NEK1杂合突变,c.1813G>A(exon21,NM_001199397),p.A605T",颅脑+颈椎+腰椎MR及脑功能成像排除其他相关病变,结合典型临床表现(中老年男性,肌肉无力,远端肢体萎缩并进行性加重)诊断为ALS。
利鲁唑50 mg、每12小时1次,依达拉奉治疗6个月(前3个月:氯化钠100 ml+依达拉奉30 mg、静脉滴注、每12小时1次,每个月连用14 d,停14 d;后3个月:氯化钠100 ml+依达拉奉30 mg、静脉滴注、每12小时1次,每个月连用10 d,停20 d)。
最后1次随访时间为2023年8月6日,患者肢体已完全瘫痪,生活完全不能自理,言语需重复方可听清,夜间睡眠困难,需要额外垫加2个枕头。改良肌萎缩侧索硬化功能评分量表评分为21分,患者疾病进展速度较快。
神经科;医学遗传科


版权归中华医学会所有。本文为遵循CC-BY-NC-ND协议的开放获取文章。
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)属于一种神经退行性疾病,主要临床特征为机体无力、肌肉萎缩并进行性加重,最终因呼吸衰竭而亡[1]。我国的年发病率为1~3/10万人,平均生存年龄3~5年[2]。NEK1基因在2014年被发现,证实与ALS的发生发展相关,发病率约占ALS的3%[3]。本文报道1例NEK1基因突变所致的ALS,旨在深入了解NEK1基因突变患者的临床特征和临床转归。
患者,男,59岁。因"四肢无力半年余,构音障碍1个月"入院。患者半年余前新冠病毒疫苗后出现双手指无力,表现为持物不稳,并逐渐向上肢近端进展,伴感觉麻木,表现为"蚁行感",当时未予重视。2个月前症状逐渐加重,并出现双下肢麻木、乏力,左下肢明显,曾因下肢无力摔倒2次,伴脚踩棉花感,伴双下肢肌肉萎缩。1个月前患者出现构音障碍,行走不便较前加重,伴饮水呛咳,无视物模糊,无吞咽困难,无肢体抽搐及不自主运动,无大小便障碍。2022年5月19日外院就诊,查颈椎MRI示"(1)颈椎骨质增生;(2)颈椎间盘变性;(3)寰枢关节后缘稍突起,增厚韧带?伴相应椎管变窄、颈髓前缘稍受压";具体诊疗不详。现患者为求进一步诊治,至本院就诊,门诊拟"运动神经元病"收入我科。自发病以来,病人精神状态一般,体力情况一般,食欲食量一般,睡眠情况一般,体重无明显变化,大便正常,小便正常。
患者2014年因外伤致右侧踝关节韧带撕裂,行手术治疗,具体不详;无高血压、糖尿病、冠心病等慢性病史;无痢疾、疟疾、病毒性肝炎及结核等传染病史。预防接种史不详。无输血史,无药物过敏史。适龄结婚,配偶身体健康,育有2女1儿,均体健。否认家族性遗传病史,否认家族性肿瘤病史。
体格检查:一般情况无明显障碍。构音低沉,咽反射对称存在,伸舌居中,可见舌肌萎缩及纤颤,余颅神经检查未见异常。运动系统四肢末梢肌容积减少,肌张力减低,肌力:屈颈5级,伸颈5级;肩内收5−级,肩外展5−级;屈腕左3−级,右4−级;伸腕左3−级,右4−级;伸指左2级,右4级;屈指左3级,右4级;握力左3−级,右4级;屈髋5级,伸髋5级;腿内收5级,外展5级;伸膝5−级,屈膝5−级;足跖屈5−级,足背伸5−级。闭目难立征(−)。感觉系统未见异常。反射:双侧腹壁反射对称存在,肱二、三头肌腱反射+++,胸大肌反射阳性,桡骨膜反射+++,膝反射+++,跟腱反射+++,Hoffmann征左侧(+),右侧(−),双侧Babinski征(−),双划征(+)。脑膜刺激征及自主神经检查未见异常。
肾功:尿酸UA 457 μmol/L,肾小球滤过率86.00 ml/min,肌酐CR 85 μmol/L。
血脂:总胆固醇3.72 mmol/L,高密度脂蛋白胆固醇0.78 mmol/L,低密度脂蛋白胆固醇2.02 mmol/L,甘油三酯4.04 mmol/L;降钙素原0.062 ng/ml。微量元素全套未见异常。血常规、离子六项、空腹血糖、糖化血红蛋白、SAA、心肌酶谱、β2-微球蛋白、同型半胱氨酸、尿常规未见明显异常。遗传代谢病氨基酸和酰基肉碱谱(血+尿)均无异常。
颈椎MRI:(1)颈椎骨质增生;(2)颈椎间盘变性;(3)寰枢关节后缘稍突起,增厚韧带?伴相应椎管变窄、颈髓前缘稍受压。常规心电图检查:(1)窦性心律;(2)正常心电图。胸部正侧位片:(1)右侧第8后肋重叠处结节影,考虑肺内结节与骨岛鉴别;(2)主动脉硬化。强迫振荡肺功能检查未见异常。肿瘤及副肿瘤相关:肿瘤标志物均阴性。
免疫相关:ANA、自身免疫十四项、血清免疫固定电泳、风湿四项、抗磷脂抗体、血管炎二项、血沉等未见明显异常。
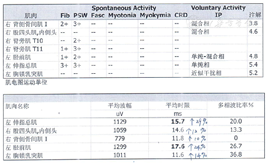
脑脊液:常规未见明显异常。生化:氯离子130.3 mmol/L;脑脊液细胞学检测未见异常。脑脊液+血清寡克隆免疫分析未见明显异常。脑功能成像(DWI,SWI,DTI):(1)DTI未见明显异常;(2)DWI及SWI未见明显异常。肌电图检查:上、下肢及胸旁肌神经源性,检查结果详见图1。
全外显子检测结果见NEK1基因突变c.1813G>A(p.A605T)。因没有对患者家属进行家系验证,所以无法对患者的突变位点来源进行分析。
四肢无力,肌肉萎缩,肌张力减低,腱反射活跃,Hoffmann征左侧(+),双划征(+),感觉、自主神经系统检查未见异常,定位于上、下运动神经元。
中老年男性,隐匿起病,慢性病程,呈进行性加重。辅助检查感染、免疫、副肿瘤等相关指标等未见异常,全外显子检测结果见NEK1基因突变c.1813G>A(p.A605T),定性为神经变性病。综合临床-电生理-病理-基因检测结果,结合EI-Escorial诊断标准,确诊ALS。
(1)颈椎病:颈椎病压迫脊髓时可致下肢腱反射亢进、双侧病理反射阳性等上、下运动神经元病变的症状和体征,但颈椎病常有感觉障碍、括约肌障碍,无延髓麻痹表象,患者外院MRI虽然提示颈椎骨质增生、颈椎间盘变性,但自主神经功能正常,肌电图示上、下肢及胸旁肌神经源性损害,排除颈椎病。
(2)多灶性运动神经病:是一种以运动神经受累为主的慢性多发性单神经病,是少见的脱髓鞘性周围神经病。其临床表现为进行性非对称性肢体无力,以远端受累为主,电生理特征是在运动神经上存在传导阻滞。本例患者有肢体无力,远端起病,远端受累更重,但患者体格检查有上运动神经元受累的表现,下肢腱反射+++,肌电图神经传导速度正常、节段传导速度未见传导阻滞,血和脑脊液神经节苷脂抗体阴性,排除该病。
(3)肯尼迪病:主要见于中年男性,表现为缓慢进展的延髓损害和肢体肌肉无力、萎缩、束颤、可有构音不全和吞咽困难;多有雄激素不足的表现,如男性乳房女性化,睾丸萎缩和阳痿等。本例患者有言语含糊,舌肌萎缩,肢体肌肉的进行性无力及萎缩,但肯尼迪病多表现为下运动神经元受累的表现,本例患者有上运动神经元受累表现,体格检查未见明显发际线后移及男性乳房女性化,性激素六项未见异常,基因筛查结果不支持,排除该病。
利鲁唑50 mg、每12小时1次,依达拉奉治疗6个月(前3个月:氯化钠100 ml+依达拉奉30 mg、静脉滴注、每12小时1次,每个月连用14 d;后3个月:每个月连用10 d)。同时康复训练及营养支持治疗。
最后1次随访时间为2023年8月6日,患者肢体已完全瘫痪,生活完全不能自理,言语需重复方可听清,夜间睡眠困难,需要额外垫加2个枕头。改良ALS功能评分量表评分为21分,患者疾病进展速度较快。
NEK1全称NIMA related kinase 1,属于高度保守的NIMA激酶家族,参与细胞物质运输和细胞周期进程及DNA的损伤修复[4]。文献表明,NEK1缺失突变所致的ALS患者有以手部无力起病的临床特点,且疾病进展较为迅速,缺失突变的患者相比错义突变的患者预后差,生存时间短,部分患者在起病1年时间内迅速出现延髓症状,而且似乎患者越早出现累及延髓的症状,患者的预后越差[5,6]。此次报道的患者以左手无力起病,在起病1年内即出现构音障碍等累及延髓的症状出现,但本例患者的突变位点属于错义突变,说明以手部无力起病的特点不仅限于缺失突变,可能是NEK1基因突变的临床特点之一,对于这一部分患者的突变位点进行了初步分析,并未发现在蛋白结构上存在分布位置集中的倾向,目前还无法解释NEK1突变尤其缺失突变所致的ALS患者大部分会出现以手部无力起病的特点。
虽然NEK1被认为是与ALS相关的基因之一,但是部分NEK1基因突变患者也可能出现其他疾病[7]。我们收集了2019—2023年在本院神经内科就诊并通过基因筛查明确存在NEK1突变的患者,发现4位患者确诊为其他神经肌肉疾病,均出现肌体无力和肌肉萎缩等早期症状相似、体征没有特异性的情况,NEK1突变类型属于杂合突变,结合临床表现、电生理、病理及其他实验室检查后排除ALS诊断,出院随访后发现进展较为缓慢,预后较ALS好。临床上累及下运动神经元的疾病常见有ALS和肯尼迪病,都属于慢性病程,尤其当肯尼迪病存在其他中枢神经系统受累时,虽然可以通过基因检测和血清肌酸激酶检测,但也可能出现基因检测阳性率低和激酶水平不增高的情况,这时就极易出现误诊的情况[8,9]。
最新的研究结果证实,NEK1通过调控TUBA1B和KPNB1参与正常生理功能,突变后会导致微管稳态失衡和核-胞浆运输障碍,从而导致神经元退化,这一现象也出现在其他家族性和散发性ALS早期疾病进展中,提示NEK1很有可能是治疗ALS极具潜力的靶点[10]。但目前对NEK1如何通过调控微管稳态和核-胞浆运输参与神经元有丝分裂的调节机制尚不明确,这需要对NEK1进一步研究。
从本例患者可以看出,NEK1基因突变所导致的疾病情况还存在极大的探究空间,尤其一些罕见的神经肌肉疾病,发病率低,且症状和体征没有特征性,极易误诊,给临床医师诊断带来极大的挑战。基因检测虽然作为一项非常有力的辅助手段,但是在诊断时,医师不能过分依赖,需要注重病史询问、体格检查及其他实验室检查,综合考量才能尽量减少误诊和漏诊。
陈晓丹,郑卉,蒋海山.NEK1基因突变致散发性肌萎缩侧索硬化1例[DB/OL].中国临床案例成果数据库,2023(2023-11-23).http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2023.e02852.
所有作者均声明本研究不存在利益冲突

























