
揭示遗传性球形细胞增多症(HS)红细胞膜蛋白基因突变特征。
应用二代测序技术检测2015年4月至2018年1月临床明确诊断的51例HS患者红细胞膜蛋白基因突变情况,将检出并预测为红细胞膜蛋白基因有害突变的37例患者纳入研究,分析基因突变构成、突变类型及与临床表现型的关系。
37例HS患者中,ANK1突变17例(45.9%)、SPTB突变14例(37.8%)、SLC4A1突变5例(13.5%)、ANK1突变复合SPTB突变1例(2.7%),未发现SPTA1及EPB42突变。红细胞膜蛋白基因突变类型中无义突变(36.8%)和错义突变(31.6%)最常见。在检出的38个突变位点中,34个为新发突变(89.5%)。16例HS患者进行父母基因验证,6例(37.5%)为遗传获得突变,10例(62.5%)为自发突变。HS患者外周血细胞参数与红细胞膜蛋白突变基因类型无关;轻型+中间型患者SPTB突变构成比更高,重型患者ANK1突变构成比更高,但差异无统计学意义(P=0.664)。
中国HS以ANK1和SPTB基因突变最常见,突变类型主要为错义突变和无义突变;不同HS相关基因突变与HS严重程度间无明显相关。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传性球形细胞增多症(Hereditary Spherocytosis,HS)是常见的先天性溶血性疾病,临床主要表现为贫血、黄疸及脾大,外周血涂片球形红细胞增多。HS散发于世界各地,北欧及北美人群发病率约1/2000[1],我国人群发病率约1/10万[2]。HS发病基础是基因突变导致红细胞膜骨架蛋白缺失或功能缺陷,约75%的患者以常染色体显性遗传方式继承于父母之一方,其余可为常染色体隐性遗传或通过胚系自发突变发病。目前已发现5种红细胞膜蛋白编码基因与HS发病相关,即ANK1、SPTA、SPTB、SLC4A1及EPB42基因,分别编码锚蛋白、α-血影蛋白、β-血影蛋白、带3蛋白和4.2蛋白。不同地域种群的膜蛋白缺乏谱不尽相同[1,3,4,5],美国、部分欧洲及韩国人群中锚蛋白缺乏最常见(占30%~60%),部分欧洲及南美人群中带3蛋白缺乏最常见,日本患者中则近一半为4.2蛋白缺乏。既往国内关于HS红细胞膜骨架蛋白及其基因突变谱的研究多为个案报告,且缺少基因突变与临床表现型之间的相关性分析。本研究中,我们采用二代测序技术检测51例HS患者红细胞膜蛋白基因突变情况,以揭示我国HS相关基因突变特征。
回顾性分析2015年4月至2018年1月,在中国医学科学院血液病医院连续就诊,根据临床表现、血液学检查和家族史明确诊断HS的51例患者资料。所有患者均进行了二代测序检测,将其中检出并预测为红细胞膜蛋白基因有害突变的37例患者纳入本研究。HS的诊断参照文献[6]标准,严重程度分型依据1990年Eber等[7]制定的诊断标准。纳入患者均来自不同家族,各家族无近亲关系。
留取患者静脉血液标本3~5 ml,采用DNA提取试剂盒(北京天根生化科技有限公司产品)及QIAamp DNA Mini Kit(德国Qiagen公司产品)提取有核细胞基因组DNA。根据GenCap定制试剂盒(北京MyGenostics公司产品)使用说明建立DNA文库,通过基因捕获策略选择已知HS发病相关的5个红细胞膜蛋白编码基因。生物素标记的捕获探针(80-120-mer)用于覆盖所有具有非重复区域的外显子,靶向区域平均基因覆盖度94.86%,平均测序深度为436.75×,90.14%的靶向区域覆盖度>30×,60.29%的靶向区域覆盖度>200×。应用Illumina HiSeq测序平台对富集的DNA文库进行测序,具体操作参照文献[8]方法。将测序结果与dbSNP数据库(http://www.ncbi.nlm.nih.gov/projects/SNP)、HGMD数据库(http://www.hgmd.cf.ac.uk/ac/index.php)进行比对。
对由二代测序检测到的HS相关基因突变,比对1000 Genomes及ExAc数据库中频率低于0.01,且功能预测为有害的位点进行Sanger测序确认。获得部分患者父母的基因组DNA,用于Sanger测序。对样本DNA进行特异性PCR扩增及纯化PCR产物,产物使用终止子循环测序方法在ABI PRISM 3730遗传分析仪上进行测序。通过将DNA序列与相应的GenBank(www.ncbi.nlm.nih.gov)参考序列进行比较来鉴定变异位点。基因参考序列转录本分别为NM_001142446(ANK1)、NM_001024858(SPTB)、NM_003126(SPTA1)、NM_000342(SLC4A1)、NM_000119(EPB42)。
应用MutationTaster(www.mutationtaster.org)、PRONEAN及SIFT(provean.jcvi.org)进行突变位点致病性预测,UniProt(www.uniprot.org)进行蛋白质功能结构域影响分析。
使用SPSS 22.0进行统计分析。连续变量以中位数(范围)进行描述,组间比较采用Kruskal-Wallis H检验;分类变量组间比较采用Fisher确切概率法。P<0.05为差异有统计学意义。
37例HS患者中,男13例(35.1%)、女24例(64.9%),中位年龄27(3~49)岁,中位HGB为97(56~142)g/L、中位网织红细胞比值为11.24%(3.57%~32.67%)、中位总胆红素为83.8(16.1~262.2)μmol/L。根据HS诊断时HGB及网织红细胞比值分型,轻型8例(21.6%)、中间型22例(59.5%)、重型7例(18.9%)。37例患者中24例(64.9%)伴有不同程度脾脏肿大。不同严重程度HS临床特征见表1。
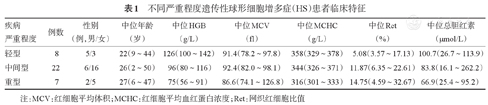
不同严重程度遗传性球形细胞增多症(HS)患者临床特征
不同严重程度遗传性球形细胞增多症(HS)患者临床特征
| 疾病严重程度 | 例数 | 性别(例,男/女) | 中位年龄(岁) | 中位HGB(g/L) | 中位MCV(fl) | 中位MCHC(g/L) | 中位Ret(%) | 中位总胆红素(μmol/L) |
|---|---|---|---|---|---|---|---|---|
| 轻型 | 8 | 5/3 | 22(9~44) | 126(100~142) | 91.4(78.2~97.8) | 358(329~378) | 5.08(3.57~17.13) | 100.7(26.7~113.9) |
| 中间型 | 22 | 6/16 | 26(2~50) | 96(80~116) | 92.4(82.0~98.1) | 344(326~371) | 11.87(6.35~22.61) | 83.8(16.1~262.2) |
| 重型 | 7 | 2/5 | 27(6~47) | 75(56~91) | 86.6(74.1~126.8) | 316(301~333) | 14.75(4.59~32.67) | 66.9(25.4~95.2) |
注:MCV:红细胞平均体积;MCHC:红细胞平均血红蛋白浓度;Ret:网织红细胞比值
37例HS患者检出红细胞膜蛋白突变基因分别涉及ANK1、SPTB及SLC4A1,均为杂合突变,未检测到SPTA1及EPB42基因突变(表2)。其中ANK1突变17例(45.9%),SPTB突变14例(37.8%),SLC4A1突变5例(13.5%),ANK1突变复合SPTB突变1例(2.7%)。
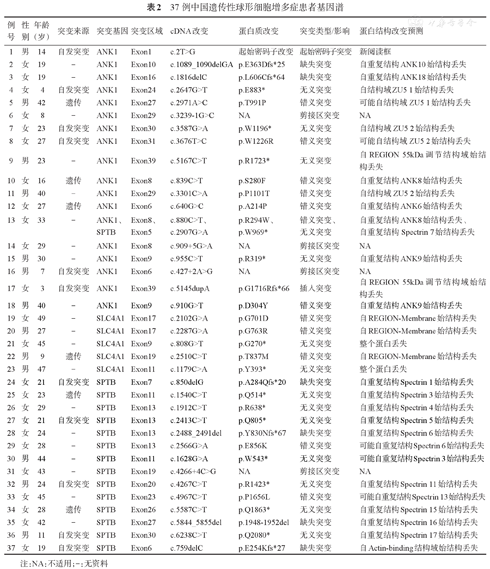
37例中国遗传性球形细胞增多症患者基因谱
37例中国遗传性球形细胞增多症患者基因谱
| 例号 | 性别 | 年龄(岁) | 突变来源 | 突变基因突变区域 | cDNA改变 | 蛋白质改变 | 突变类型/影响 | 蛋白结构改变预测 | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 男 | 14 | 自发突变 | ANK1 | Exon1 | c.2T>G | 起始密码子改变 | 起始密码子突变 | 新阅读框 |
| 2 | 女 | 19 | - | ANK1 | Exon10 | c.1089_1090delGA | p.E363Dfs*25 | 缺失突变 | 自重复结构ANK10始结构丢失 |
| 3 | 女 | 19 | - | ANK1 | Exon16 | c.1816delC | p.L606Cfs*64 | 缺失突变 | 自重复结构ANK18始结构丢失 |
| 4 | 女 | 4 | 自发突变 | ANK1 | Exon24 | c.2647G>T | p.E883* | 无义突变 | 自结构域ZU5 1始结构丢失 |
| 5 | 男 | 42 | 遗传 | ANK1 | Exon27 | c.2971A>C | p.T991P | 错义突变 | 可能自结构域ZU5 1始结构丢失 |
| 6 | 女 | 8 | - | ANK1 | Exon29 | c.3239-1G>C | NA | 剪接区突变 | NA |
| 7 | 女 | 23 | 自发突变 | ANK1 | Exon30 | c.3587G>A | p.W1196* | 无义突变 | 自结构域ZU5 2始结构丢失 |
| 8 | 女 | 27 | 自发突变 | ANK1 | Exon31 | c.3676T>C | p.W1226R | 错义突变 | 可能自结构域ZU5 2始结构丢失 |
| 9 | 男 | 23 | - | ANK1 | Exon39 | c.5167C>T | p.R1723* | 无义突变 | 自REGION 55kDa调节结构域始结构丢失 |
| 10 | 女 | 16 | 遗传 | ANK1 | Exon8 | c.839C>T | p.S280F | 错义突变 | 自重复结构ANK8始结构丢失 |
| 11 | 男 | 40 | - | ANK1 | Exon29 | c.3301C>A | p.P1101T | 错义突变 | 自结构域ZU5 2始结构丢失 |
| 12 | 女 | 27 | 遗传 | ANK1 | Exon6 | c.640G>C | p.A214P | 错义突变 | 自重复结构ANK6始结构丢失 |
| 13 | 女 | 33 | - | ANK1、 | Exon8、 | c.880C>T、 | p.R294W、 | 错义突变、 | 自重复结构ANK8始结构丢失、 |
| SPTB | Exon5 | c.2907G>A | p.W969* | 无义突变 | 自重复结构Spectrin 7始结构丢失 | ||||
| 14 | 女 | 29 | - | ANK1 | Exon8 | c.909+5G>A | NA | 剪接区突变 | NA |
| 15 | 男 | 30 | - | ANK1 | Exon9 | c.955C>T | p.R319* | 无义突变 | 自重复结构ANK9始结构丢失 |
| 16 | 男 | 7 | 自发突变 | ANK1 | Exon6 | c.427+2A>G | NA | 剪接区突变 | NA |
| 17 | 女 | 3 | 自发突变 | ANK1 | Exon39 | c.5145dupA | p.G1716Rfs*66 | 插入突变 | 自REGION 55kDa调节结构域始结构丢失 |
| 18 | 男 | 40 | - | ANK1 | Exon9 | c.910G>T | p.D304Y | 错义突变 | 自重复结构ANK9始结构丢失 |
| 19 | 女 | 49 | - | SLC4A1 | Exon17 | c.2102G>A | p.G701D | 错义突变 | 自REGION-Membrane始结构丢失 |
| 20 | 男 | 27 | - | SLC4A1 | Exon17 | c.2287G>A | p.G763R | 错义突变 | 自REGION-Membrane始结构丢失 |
| 21 | 女 | 45 | - | SLC4A1 | Exon9 | c.808G>T | p.G270* | 无义突变 | 整个蛋白丢失 |
| 22 | 男 | 9 | 遗传 | SLC4A1 | Exon19 | c.2510C>T | p.T837M | 错义突变 | 自REGION-Membrane始结构丢失 |
| 23 | 男 | 47 | - | SLC4A1 | Exon11 | c.1179C>A | p.Y393* | 无义突变 | 整个蛋白丢失 |
| 24 | 女 | 21 | 自发突变 | SPTB | Exon7 | c.850delG | p.A284Qfs*20 | 缺失突变 | 自重复结构Spectrin 1始结构丢失 |
| 25 | 女 | 23 | 遗传 | SPTB | Exon11 | c.1540C>T | p.Q514* | 无义突变 | 自重复结构Spectrin 3始结构丢失 |
| 26 | 女 | 29 | - | SPTB | Exon13 | c.1912C>T | p.R638* | 无义突变 | 自重复结构Spectrin 4始结构丢失 |
| 27 | 女 | 21 | 自发突变 | SPTB | Exon13 | c.2413C>T | p.Q805* | 无义突变 | 自重复结构Spectrin 5始结构丢失 |
| 28 | 女 | 24 | - | SPTB | Exon13 | c.2488_2491del | p.Y830Nfs*67 | 缺失突变 | 自重复结构Spectrin 6始结构丢失 |
| 29 | 女 | 28 | - | SPTB | Exon13 | c.2566G>A | p.E856K | 错义突变 | 可能自重复结构Spectrin 6始结构丢失 |
| 30 | 男 | 44 | - | SPTB | Exon11 | c.1628G>A | p.W543* | 无义突变 | 可能自重复结构Spectrin 3始结构丢失 |
| 31 | 女 | 43 | - | SPTB | Exon19 | c.4266+4C>G | NA | 剪接区突变 | NA |
| 32 | 男 | 24 | 自发突变 | SPTB | Exon20 | c.4267C>T | p.R1423* | 无义突变 | 自重复结构Spectrin 11始结构丢失 |
| 33 | 女 | 45 | - | SPTB | Exon23 | c.4967C>T | p.P1656L | 错义突变 | 可能自重复结构Spectrin 13始结构丢失 |
| 34 | 女 | 28 | 遗传 | SPTB | Exon26 | c.5587C>T | p.Q1863* | 无义突变 | 自重复结构Spectrin 15始结构丢失 |
| 35 | 女 | 42 | - | SPTB | Exon27 | c.5844_5855del | p.1948-1952del | 缺失突变 | 自重复结构Spectrin 16始结构丢失 |
| 36 | 男 | 11 | 自发突变 | SPTB | Exon30 | c.6238C>T | p.Q2080* | 无义突变 | 自重复结构Spectrin 17始结构丢失 |
| 37 | 女 | 19 | 自发突变 | SPTB | Exon6 | c.759delC | p.E254Kfs*27 | 缺失突变 | 自Actin-binding结构域始结构丢失 |
注:NA:不适用;-:无资料
38个红细胞膜蛋白基因致病突变中,无义突变(14/38,36.8%)和错义突变(12/38,31.6%)最常见,其后依次为缺失突变(6/38,15.8%)、剪接区突变(4/38,10.5%)、插入突变(1/38,2.6%)和起始密码子突变(1/38,2.6%)。18个ANK1突变中,错义突变7个(38.9%)、无义突变4个(22.2%)、剪接区突变3个(16.7%)、缺失突变2个(11.1%)、插入突变及起始密码子突变1个(5.6%);15个SPTB突变中,无义突变8个(53.3%)、缺失突变4个(26.7%)、错义突变2个(13.3%)、剪接区突变1个(6.7%);5个SLC4A1突变中,错义突变3个(60.0%)、无义突变2个(40.0%)。
通过与HGMD数据库(http://www.hgmd.cf.ac.uk)中已报道HS相关基因突变位点进行对比,34个突变未见报道(89.5%),而4例患者(例19、22、25及26)的膜蛋白基因突变类型已有报道。
应用Sanger测序对16例HS患者的父母进行基因突变检测。6例(37.5%)患者的父/母可检测到相同突变;10例(62.5%)患者的亲代未检测到相同突变,为红细胞膜蛋白基因自发突变。
对突变位点进行结局预测显示,大多数基因(26/38,68.4%)突变导致提前出现转录终止密码子,生成截短肽链,致使蛋白被降解或不能形成功能蛋白。其中14例患者为点突变后获得终止密码子;7例发生核苷酸缺失或插入,致框移突变并提前出现终止密码子;4例剪接区突变可能致使mRNA剪接过程改变生成截短蛋白;1例基因起始密码子突变致使读码框改变,不能转录翻译相应蛋白。另12例患者点突变致单个氨基酸改变,预测可能致使相应功能域后肽链缺失。
18个ANK1突变中,7个累及锚蛋白膜结合结构域、5个累及ZU5亚功能域、2个累及调节区域。15个SPTB突变中,13个累及β亚单位的重复单位、1个累及肌钙蛋白结合结构域。5个SLC4A1突变中,2个无义突变致带3蛋白缺失,3个错义突变累及跨膜区域。
检出的3种常见红细胞膜蛋白编码基因ANK1、SPTB及SLC4A1突变的HS患者外周血细胞参数见表3,不同的膜蛋白编码基因突变患者其外周血细胞参数相近(P值均>0.05)。比较最常见的ANK1和SPTB基因突变HS的疾病严重程度(未纳入例13 ANK1、SPTB复合突变患者),结果显示17例ANK1突变者中轻型+中间型13例(76.5%),重型4例(23.5%);14例SPTB突变者中轻型+中间型12例(85.7%),重型2例(14.3%)。轻型+中间型患者SPTB突变构成比更高,重型患者ANK1突变构成比更高,但差异无统计学意义(P=0.664)。ANK1突变累及结合结构域、ZU5功能域及调节区域不同锚蛋白结构域者,中位HGB水平分别为104(84~116)、88(73~129)及92(78~105)g/L。比较不同突变类型与外周血细胞参数的关系,结果示错义突变、无义突变、移码突变及其他突变类型外周血细胞参数之间差异无统计学意义(表4)。
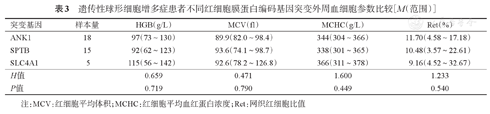
遗传性球形细胞增多症患者不同红细胞膜蛋白编码基因突变外周血细胞参数比较[M(范围)]
遗传性球形细胞增多症患者不同红细胞膜蛋白编码基因突变外周血细胞参数比较[M(范围)]
| 突变基因 | 样本量 | HGB(g/L) | MCV(fl) | MCHC(g/L) | Ret(%) |
|---|---|---|---|---|---|
| ANK1 | 18 | 97(73~130) | 89.9(82.0~98.4) | 344(304~366) | 11.70(4.58~17.18) |
| SPTB | 15 | 92(62~123) | 93.6(74.1~98.7) | 338(301~365) | 10.48(3.57~22.61) |
| SLC4A1 | 5 | 115(56~142) | 92.6(78.2~126.8) | 366(311~378) | 9.16(4.52~32.67) |
| H值 | 0.659 | 0.471 | 1.600 | 1.233 | |
| P值 | 0.719 | 0.790 | 0.449 | 0.540 |
注:MCV:红细胞平均体积;MCHC:红细胞平均血红蛋白浓度;Ret:网织红细胞比值
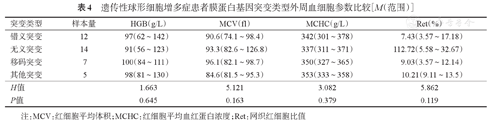
遗传性球形细胞增多症患者膜蛋白基因突变类型外周血细胞参数比较[M(范围)]
遗传性球形细胞增多症患者膜蛋白基因突变类型外周血细胞参数比较[M(范围)]
| 突变类型 | 样本量 | HGB(g/L) | MCV(fl) | MCHC(g/L) | Ret(%) |
|---|---|---|---|---|---|
| 错义突变 | 12 | 97(62~142) | 90.6(74.1~98.4) | 342(301~378) | 7.43(3.57~17.18) |
| 无义突变 | 14 | 91(56~123) | 93.3(82.6~126.8) | 337(311~371) | 112.72(5.58~32.67) |
| 移码突变 | 7 | 100(84~111) | 96.1(82.1~98.7) | 350(327~365) | 9.03(3.57~12.14) |
| 其他突变 | 5 | 98(81~130) | 84.6(81.5~95.3) | 353(333~358) | 10.21(9.11~13.5) |
| H值 | 1.663 | 5.121 | 3.082 | 5.862 | |
| P值 | 0.645 | 0.163 | 0.379 | 0.119 |
注:MCV:红细胞平均体积;MCHC:红细胞平均血红蛋白浓度;Ret:网织红细胞比值
HS是由编码红细胞膜骨架蛋白基因突变所致的先天性溶血性疾病,已知的致病基因涉及ANK1、SPTB、SPTA1、SLC4A1和EPB42[1]。多数患者呈常染色体显性遗传模式。本研究中的16例接受亲代验证的患者中,自发基因突变者高达62.5%,这可能与我院家族史阴性疑难患者就诊比例较高有关。ANK1是最常见的致病基因,约占50%;其次是血影蛋白基因(SPTB突变约占20%、SPTA1突变约占5%);SLC4A1突变约占15%;EPB42突变约占10%[4,5,9,10,11]。不同国家及种族基因突变发生率有所差异。Nakanishi等[12]报道日本HS患者中约1/3存在ANK1基因突变;Park等[13]对25例韩国HS患者进行基因突变分析,52%为ANK1杂合突变、48%为SPTB杂合突变。而美国及欧洲HS患者中25%为SPTB杂合突变所致[14]。我们对37例中国HS患者红细胞膜蛋白基因突变研究结果显示ANK1突变占45.9%、SPTB突变占37.8%,与日本、韩国报告结果相近,提示ANK1和SPTB为亚洲人群HS最常见致病基因。
锚蛋白是由ANK1编码的红细胞膜垂直方向膜蛋白,其连接红细胞穿膜蛋白带3蛋白及血影蛋白,以维持红细胞膜的稳定性及变形性。已在HS中共发现80余种ANK1基因致病突变,以移码突变及无义突变类型较为多见,此类突变导致转录产物不稳定,其mRNA被降解或是产生截短型缺陷锚蛋白分子,另外也有低频的错义突变及剪接区突变发生[5,9]。在ANK1突变中,约75%突变为遗传性突变,但各家系间突变位点各不相同,未发现热点突变位点或区域[9,11,12,13,15,16,17,18,19,20,21]。本研究显示,HS患者中近半数突变基因为ANK1,亲代验证结果显示其中仅3例遗传自亲代,6例为自发突变。检出的18个ANK1突变位点均无文献报道,为新发突变,以错义突变和无义突变为主,分别占38.9%和22.2%。结合HS患者临床症状及蛋白功能预测,推定这些新发突变均为致病突变,并且锚蛋白3个功能域均有累及。
血影蛋白是红细胞膜骨架网络中重要的组成成分,其通过与锚蛋白相结合而固定于细胞膜。SPTA1和SPTB分别编码α和β亚单位,两者组成同源二聚体即为血影蛋白。其中β亚单位中血影蛋白重复区14和15为锚蛋白结合区。HS相关血影蛋白基因突变中,编码β亚单位的SPTB突变更为常见,其多为常染色体显性遗传;而编码α亚单位的SPTA1突变所致HS较为少见,多为常染色体隐性遗传方式。既往研究发现,HS中SPTB突变主要为移码突变、无义突变、剪接区突变及起始密码子突变所致的无效突变,致表达蛋白缺失或是功能缺陷。本组患者的血影蛋白基因突变主要累及SPTB,其中半数遗传自亲代,未检测到SPTA1突变;15个SPTB突变位点中,12个为新发突变,主要累及血影蛋白重复单位致其锚蛋白结合能力丧失。
带3蛋白为红细胞膜含量最多且最重要的跨膜离子通道蛋白。文献报道的81个HS相关SLC4A1突变中,错义突变、移码突变和剪接区突变最为常见;其基因突变亦呈遗传特性,各家系间突变位点少见重叠,未见突变热点[9,22,23,24,25,26,27,28,29]。本组病例中SLC4A1突变占13.5%(5/37),包括3例错义突变和2例无义突变,主要影响带3蛋白跨膜区域或致完整带3蛋白缺失。其中SLC4A1 p.T837M、p.G701D突变曾有报道;另外3例SLC4A1突变均为新发突变。
4.2蛋白是由EPB42基因编码的转谷氨酰胺酶蛋白家族成员,其功能主要为稳定血影蛋白-肌动蛋白-锚蛋白与带3蛋白的联结。4.2蛋白缺乏者多为常染色体隐性遗传,日本人群中多见,其他人群中少有报道,发病率<5%[28,29]。本组37例中国HS患者中未发现EPB42突变。
不同致病基因的突变或可导致不同临床表现型。韩国研究报告显示,ANK1突变累及血影蛋白结合域的患者HGB水平明显低于累及膜结合域或调节域者[13],但不同严重程度HS患者ANK1、SPTB突变发生率并无明显差异。我们对HS相关基因突变型与临床表现型进行分析未发现不同膜蛋白基因突变患者外周血细胞参数存在差异;在ANK1突变中,不同蛋白编码结构域突变患者其HGB水平也无明显差异。我们认为这可能与本研究样本数相对较小,而影响HS严重程度因素较多有关。除红细胞膜缺陷外,骨髓造血代偿能力和是否伴发造血原料缺乏、感染等诱发病情加重因素,也可影响对HS严重程度的判断。
本研究结果显示我国HS红细胞膜骨架蛋白突变基因约86%为ANK1和SPTB突变,SLC4A1突变仅占13.5%,未发现SPTA1及EPB42突变;膜骨架蛋白基因缺乏突变热点,且新发突变非常普遍;HS基因以无义突变、错义突变为主,不同突变基因和突变类型患者外周血细胞参数之间无明显差异。

























