
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
混合表型急性白血病(MPAL)是一种少见类型的白血病,表达一种以上系列特异性抗原。根据WHO 2016分类标准,MPAL分为五种亚型:MPAL伴t(9;22)(q34;q11.2);MPAL伴MLL重排;MPAL,B/髓系,非特指型(NOS);MPAL,T/髓系,NOS;MPAL罕见型,NOS。我们收集了25例成人MPAL患者临床资料,对其细胞遗传学及基因学特征、治疗及预后进行了回顾性分析,报道如下。
收集河南省人民医院2011年1月至2018年1月收治的资料完整的25例成人MPAL患者,诊断符合2016 WHO血液肿瘤分类标准,排除急性髓系白血病(AML)伴重现性遗传学异常、AML伴骨髓增生异常相关改变(AML-MRC)、治疗相关AML、慢性髓性白血病急变期。男16例,女9例,中位年龄42(20~71)岁。
采用美国BD公司生产的BD FACSCablibur流式细胞仪进行检测,非谱系抗体为CD34、CD38、CD123、HLA-DR;髓系抗体为MPO、CD117、CD33、CD13、CD11b、CD11c、CD14、CD15、CD64;淋系抗体为CD3、CD5、CD7、CD2、CD4、CD8、CD10、CD19、CD20、CD22、CD9、CD79a、CD56。膜抗原阳性率≥20%定义为阳性,胞质抗原阳性率≥10%定义为阳性。
骨髓细胞经24 h培养常规制片,应用RHG显带技术进行染色体核型分析,每例分析10~20个分裂象,异常克隆根据《人类细胞遗传学国际命名体制(ISCN)》(2013)进行描述,复杂核型定义为≥3种染色体异常。
应用RT-PCR方法进行融合基因检测,融合基因包括急性淋巴细胞白血病(ALL)和AML常见的如BCR-ABL、AML1-ETO、CBFβ-MYH11、MLL和PML-RARα相关等43种融合基因。采用Illumina测序平台对2015至2018年收治的11例患者的ASXL1、IDH1、DNMT3A、SF3B1D等在内的82个白血病相关基因进行测序,平均基因覆盖率100%,平均测序深度2 000×,99%的目标区域测序深度超过20×,检测范围包括基因编码区点突变、小片段插入、缺失等突变类型。
诱导方案:髓系方案:MEA(米托蒽醌、依托泊苷、阿糖胞苷)、HAA(高三尖杉酯碱、阿克拉霉素、阿糖胞苷)、D/IA(柔红霉素/去甲氧柔红霉素、阿糖胞苷);淋髓兼顾方案:DOAP(柔红霉素、长春新碱、阿糖胞苷、泼尼松);淋系方案:VDCLP(长春新碱、柔红霉素、环磷酰胺、左旋门冬酰胺酶、泼尼松/地塞米松)。Ph染色体阳性者化疗同时加用伊马替尼400 mg/d口服治疗。缓解后继续巩固治疗,有合适供者的进行异基因造血干细胞移植。
主要采用门诊随访及电话随访方式,随访截止日期为2018年6月30日。
采用SPSS 22.0软件进行统计学分析。计量资料符合正态分布的用平均值描述,组间比较采用t检验。若计量资料呈非正态分布,用中位数描述,组间分布比较采用Mann-Whitney U检验。定性资料采用卡方检测。Kaplan-Meier法进行生存分析,总生存(OS)时间指疾病确诊至死亡的时间或随访终点,组间比较采用Log-rank检验。检验水准α=0.05,双侧检验。P<0.05为差异有统计学意义。
初诊时临床表现为感染、发热14例,乏力9例,肝脾淋巴结肿大6例,皮肤紫癜、瘀斑、齿龈出血4例,下肢静脉血栓1例。中位WBC 34.1(1.1~380.3)×109/L,HGB 74(41~123)g/L,PLT 35(6~329)×109/L。
17例为B/髓系型,7例为T/髓系型,1例为B/T型。在大多数B/髓系型患者中表达MPO、CD13、CD33、CD10、CD19、CD79a、CD34、HLA-DR、CD38、CD123。在大多数T/髓系型患者中,表达MPO、CD13、CD33、CD117、CD3、CD7、CD34、HLA-DR、CD38、CD123。1例T/B型,表达CD38、CD123、CD13、CD19、CD10、CD79a、CD3、CD7。具体表型特征见表1。B/髓系型与T/髓系型在WBC、PLT、HGB、年龄、性别方面差异无统计学意义(P值均>0.05)(表2)。
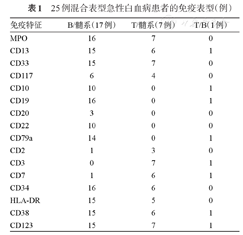
25例混合表型急性白血病患者的免疫表型(例)
25例混合表型急性白血病患者的免疫表型(例)
| 免疫特征 | B/髓系(17例) | T/髓系(7例) | T/B(1例) |
|---|---|---|---|
| MPO | 16 | 7 | 0 |
| CD13 | 15 | 6 | 1 |
| CD33 | 15 | 7 | 0 |
| CD117 | 6 | 4 | 0 |
| CD10 | 10 | 0 | 1 |
| CD19 | 16 | 0 | 1 |
| CD20 | 3 | 0 | 0 |
| CD22 | 10 | 0 | 0 |
| CD79a | 14 | 0 | 1 |
| CD2 | 1 | 3 | 0 |
| CD3 | 0 | 7 | 1 |
| CD7 | 1 | 6 | 1 |
| CD34 | 16 | 6 | 0 |
| HLA-DR | 15 | 5 | 0 |
| CD38 | 15 | 6 | 1 |
| CD123 | 15 | 7 | 1 |
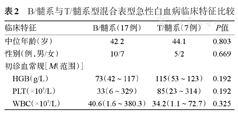
B/髓系与T/髓系型混合表型急性白血病临床特征比较
B/髓系与T/髓系型混合表型急性白血病临床特征比较
| 临床特征 | B/髓系(17例) | T/髓系(7例) | P值 | |
|---|---|---|---|---|
| 中位年龄(岁) | 42.2 | 44.1 | 0.803 | |
| 性别(例,男/女) | 10/7 | 5/2 | 0.669 | |
| 初诊血常规[M(范围)] | ||||
| HGB(g/L) | 73(42~117) | 115(53~123) | 0.192 | |
| PLT(×109/L) | 33(6~329) | 85(23~314) | 0.192 | |
| WBC(×109/L) | 40.6(1.6~380.3) | 34.2(1.1~72.7) | 0.325 | |
除1例未见分裂象外,24例核型分析成功:正常核型8例,Ph染色体阳性6例(1例附加16号染色体异常),Y染色体缺失2例,复杂核型1例,add(1)(q?)、+8、+12、add(9)(q34)、dup(1)(q24q44)各1例,其他异常2例。
6例BCR-ABL基因阳性,与染色体核型一致,4例为P190阳性,2例为P210阳性。
排除基因多态性和同义突变后,11例患者中8例检出基因突变。在ATRX、DNMT3A、EZH2、FBXW7、IKZF1、JAK1、KMT2A、NOTCH1、NRAS、PHF6、RUNX1、SF3B1、TET2、WT1等14个基因共检出16个位点突变,具体见表3。唯一重复出现的是RUNX1基因突变,1例PHF6基因检出两个位点突变,基因突变最多的患者存在4种基因突变,5例患者中至少存在2种基因突变。

11例混合表型急性白血病二代测序结果
11例混合表型急性白血病二代测序结果
| 例号 | 性别 | 年龄(岁) | 表型 | 核型 | 突变基因 |
|---|---|---|---|---|---|
| 1 | 男 | 41 | B/髓系 | 45,X,-Y[20] | 未检出 |
| 2 | 男 | 31 | B/髓系 | 未见分裂象 | 未检出 |
| 3 | 女 | 35 | B/髓系 | 46,XX,t(9;22)(q34;q11)[10]/46,XX[7] | ATRX |
| 4 | 男 | 23 | B/髓系 | 46,XY[15] | NRAS、RUNX1 |
| 5 | 男 | 71 | B/髓系 | 46,XY[10] | IKZF1、EZH2 |
| 6 | 女 | 58 | B/髓系 | 46,XX,t(9;22)(q34;q11),add(16)(q?)[20] | FBXW7 |
| 7 | 男 | 43 | B/髓系 | 45,X,-Y[20] | DNMT3A、RUNX1、TET2 |
| 8 | 男 | 45 | T/髓系 | 46,XY,t(9;22)(q34:q11)[20] | 未检出 |
| 9 | 男 | 25 | T/髓系 | 分裂象均为多倍体,可见6q+,其他异常无法分析 | JAK1、NOTCH1、PHF6 |
| 10 | 女 | 67 | T/髓系 | 46,XX[15] | WT1、KMT2A |
| 11 | 男 | 31 | T/B | 45,XY,del(9)(p13),add(14)(p11),-20[20] | SF3B1 |
除1例化疗前死亡外,余24例接受化疗。总缓解率为63%(15/24),髓系方案缓解率为54%(7/13)、淋髓兼顾方案为71%(5/7)、淋系方案为75%(3/4),三种化疗方案缓解率相比差异无统计学意义(P=0.623)。中位OS时间为10.1个月,B/髓系型与T/髓系型OS时间差异无统计学意义(P=0.581)。Ph阳性组缓解率50%(3/6),Ph阴性组缓解率67%(12/18),差异无统计学意义(P=0.635);Ph阳性组中位OS时间为7.1个月,Ph阴性组中位OS时间为12.0个月,差异无统计学意义(P=0.103)。15例缓解患者中,5例接受了异基因造血干细胞移植:1例Ph阳性患者死于复发,1例死于小细胞肺癌,3例无病生存。10例未移植患者均死亡,8例死于复发,2例死于化疗后感染。缓解后移植组与未移植组OS差异有统计学意义(P=0.003)。
MPAL是一种罕见的急性白血病,占所有急性白血病的2%~3%[1]。在大多数报道中,MPAL年龄分布广,男女比为1.5∶1[2],本组病例中年龄分布、性别比例与文献相符。在本研究中,MPAL患者初诊表现主要为贫血、出血、感染、白血病浸润等症状,60%以上的患者WBC>10×109/L,部分患者可出现白细胞瘀滞。本组病例最常见的免疫表型为B/髓系(68%),T/髓系(28%)次之,T/B(4%)罕见,无B/T/髓系表型,与国内外相关研究一致[1,3]。
遗传学异常在MPAL中很常见,在两项病例最多的回顾性研究中,87%[1]和64%[3]的MPAL患者检出异常核型。MPAL伴Ph阳性、MPAL伴MLL重排是WHO分类中单独的亚型。在MPAL中Ph阳性率为15%~20%,主要发生于成年人,MLL重排发生率为4%~8%,在儿童MPAL(尤其是婴儿)中更为常见[4]。在本组成人病例中67%有异常核型,Ph染色体最常见(6例,25%),未见MLL重排。在AML和ALL中,Ph染色体是预后不良的因素[5,6],Matutes等[1]研究发现Ph阳性MPAL中位OS时间最短。在本研究中Ph阳性组与Ph阴性组相比,缓解率低,生存期短,但是差异无统计学意义,考虑与样本量较少和靶向治疗改善了疗效有关。一系列研究报道了MPAL中基因突变的种类和频率,由于检测基因数目与检测手段的差异,基因突变检出率39%~94%[3, 7,8,9]。本研究中,应用二代测序检测82个相关基因,73%(8/11)可检出突变,45%(5/11)为多基因突变,涉及染色质调节(ATRX、EZH2)、DNA甲基化(DNMT3A、TET2、KMT2A)、转录因子(IKZF1、RUNX1、PHF6、WT1)、RNA剪接(SF3B1)、JAK-STAT通路(JAK1)、NOTCH通路(FBXW7、NOTCH1)、RTK-RAS通路(NRAS)。与混合免疫表型一致的是ALL型和AML型突变均可检测到,常见于ALL、鲜少在AML发生的PHF6和NOTCH1突变,NRAS、DNMT3A等髓系相关基因突变。RUNX1、DNMT3A、SF3B1、EZH2等突变在AML中是预后不良因素[5, 10]。多基因突变在AML中与高龄、高白细胞等不良预后有关[11]。研究发现伴基因突变的MPAL OS更差[8]。本研究中1例DNMT3A、RUNX1共突变患者在化疗后脑出血死亡;1例RUNX1、NRAS共突变患者与1例EZH2、IKZF1共突变患者治疗过程中始终未获得缓解;1例SF3B1突变患者治疗后获得了缓解,但是早期复发,仅生存7.5个月;而未检出基因突变的3例患者均获得了缓解且OS时间超过1年。提示我们RUNX1、DNMT3A、SF3B1、EZH2基因突变可能影响MPAL预后,但目前相关研究样本数偏少,需要多中心协作纳入更多样本评估特定突变和免疫表型之间的相关性,确定MPAL突变与预后相关性。
目前MPAL尚无标准治疗方案,临床实践中常使用AML、ALL或两系兼顾的方案诱导治疗。回顾性研究显示ALL方案较AML方案缓解率更高[12],淋髓兼顾方案与ALL方案缓解率相当[13]。最新的研究显示根据MPAL的甲基化谱分为ALL型MPAL和AML型MPAL,给予匹配的治疗方案缓解率更高[7]。在本组研究中,ALL方案缓解率最高,但是差异无统计学意义。复发是本组缓解后MPAL患者死亡的主要原因,移植组较单纯化疗组生存有优势,移植后有60%的患者获得了2年以上无病生存。
总之,MPAL是一种异质性较强的白血病类型,多伴有预后不良的细胞遗传学异常及预后较差的基因突变,治疗缓解率低,复发率高,移植是治愈的主要手段。

























