
分析Rosai-Dorfman disease(RDD)的临床病理特征,利用全基因组外显子测序探讨RDD的发病机制。
回顾性分析第二军医大学附属长海医院、长征医院2010年1月至2018年7月收治的23例RDD患者临床病理资料,并对9例患者石蜡包埋组织标本进行了全基因组外显子测序。
23例RDD患者中位年龄47(10~79)岁,19例为结外型,3例为淋巴结型,1例为混合型。所有患者均接受了手术切除病灶,19例患者中位随访24(1~67)个月,均无复发。病理形态主要表现为淋巴结窦内或结外组织中组织细胞增生伴有噬淋巴细胞现象,免疫组化示组织细胞表达S100、CD68、CD163,不表达CD1a。全基因组外显子测序发现mTOR、KMT2D和NOTCH1基因突变。
mTOR、KMT2D和NOTCH1基因突变可能参与了RDD的发病机制,其临床意义仍需要进一步研究。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Rosai-Dorfman病(RDD),又名窦组织细胞增生伴巨大淋巴结病(Sinus histiocytosis with massive lymph adenopathy,SHML),是一种少见的良性组织细胞增生性疾病。RDD的病因尚不明确,临床主要表现为淋巴结无痛性肿大或淋巴结外软组织肿块,可伴有发热、盗汗、体重减轻等。既往报道RDD与RAS、PIK3CA、TNFRSF6、MAP2K1、BRAF及ARAF突变有关。本研究中,我们总结了23例RDD患者临床资料,并对9例患者行全基因组外显子测序(whole exome sequencing,WES),现报道如下并进行文献复习。
以第二军医大学附属长海医院、长征医院2010年1月至2018年7月23例RDD患者临床病理资料为研究对象,结合患者临床资料及随访结果进行分析。
所有23例RDD患者均经外科手术切除行病理活检,固定、脱水、石蜡包埋切片,HE染色进行形态学观察,EnVision法进行免疫组化标记。一抗S100、CD68、CD163、CD1a、CD38、CD138、Kappa、Lambda、CD3、CD20、CD21、CD23等均为丹麦DAKO公司产品。其中9例患者石蜡包埋组织标本送至上海澳斯泰医学检验所行WES。采用德国Qiagen公司的QIAamp DNA FFPE Tissue Kit试剂盒提取石蜡样本DNA,以美国KAPA公司的Hyper Prep Kit Illumina Platforms文库构建试剂盒制备文库,采用美国Agilent公司的High Sensitivity DNA Kit试剂对文库进行质控。选用瑞氏Roche公司的SeqCap EZ Exome对质控后的文库进行杂交,捕获外显子区序列。美国Illumina公司HiSeq X Ten测序仪对质控合格的杂交后的文库进行双端高通量测序。
主要采用病案查询、门诊复查及电话进行随访,随访截至2018年9月1日,4例患者失访,余19例中位随访时间24(1~67)个月。
23例RDD患者中男13例,女10例,男女比为1.3∶1,中位年龄47(10~79)岁,19例为结外型,分别位于皮肤、眼眶、阴囊、气管、鼻咽部、乳腺、脊柱和腹膜,3例为淋巴结型,分别位于颈部、肠系膜和颏下淋巴结,1例为混合型,累及结肠及周围淋巴结。16例(70%)患者以发现体表肿物起病,其余患者临床表现与病变累及部位相关。所有患者出现临床症状至明确诊断的时间为2个月至3年不等。临床资料见表1。
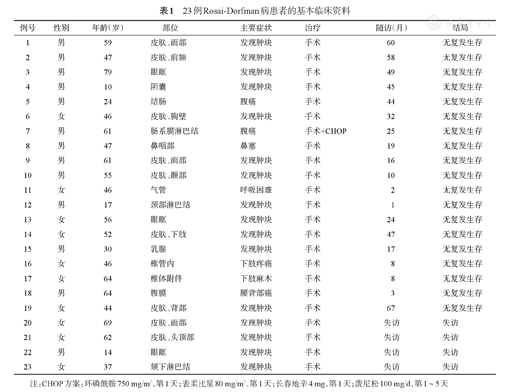
23例Rosai-Dorfman病患者的基本临床资料
23例Rosai-Dorfman病患者的基本临床资料
| 例号 | 性别 | 年龄(岁) | 部位 | 主要症状 | 治疗 | 随访(月) | 结局 |
|---|---|---|---|---|---|---|---|
| 1 | 男 | 59 | 皮肤、面部 | 发现肿块 | 手术 | 60 | 无复发生存 |
| 2 | 男 | 47 | 皮肤、前额 | 发现肿块 | 手术 | 58 | 无复发生存 |
| 3 | 男 | 79 | 眼眶 | 发现肿块 | 手术 | 49 | 无复发生存 |
| 4 | 男 | 10 | 阴囊 | 发现肿块 | 手术 | 45 | 无复发生存 |
| 5 | 男 | 24 | 结肠 | 腹痛 | 手术 | 44 | 无复发生存 |
| 6 | 女 | 46 | 皮肤、胸壁 | 发现肿块 | 手术 | 32 | 无复发生存 |
| 7 | 男 | 61 | 肠系膜淋巴结 | 腹痛 | 手术+CHOP | 25 | 无复发生存 |
| 8 | 男 | 47 | 鼻咽部 | 鼻塞 | 手术 | 19 | 无复发生存 |
| 9 | 男 | 61 | 皮肤、面部 | 发现肿块 | 手术 | 16 | 无复发生存 |
| 10 | 男 | 55 | 皮肤、颞部 | 发现肿块 | 手术 | 10 | 无复发生存 |
| 11 | 女 | 46 | 气管 | 呼吸困难 | 手术 | 2 | 无复发生存 |
| 12 | 男 | 17 | 颈部淋巴结 | 发现肿块 | 手术 | 1 | 无复发生存 |
| 13 | 女 | 56 | 眼眶 | 发现肿块 | 手术 | 24 | 无复发生存 |
| 14 | 女 | 52 | 皮肤、下肢 | 发现肿块 | 手术 | 47 | 无复发生存 |
| 15 | 男 | 30 | 乳腺 | 发现肿块 | 手术 | 17 | 无复发生存 |
| 16 | 女 | 46 | 椎管内 | 下肢疼痛 | 手术 | 8 | 无复发生存 |
| 17 | 女 | 64 | 椎体附件 | 下肢麻木 | 手术 | 8 | 无复发生存 |
| 18 | 男 | 64 | 腹膜 | 腰背部痛 | 手术 | 3 | 无复发生存 |
| 19 | 女 | 44 | 皮肤、背部 | 发现肿块 | 手术 | 67 | 无复发生存 |
| 20 | 女 | 69 | 皮肤、面部 | 发现肿块 | 手术 | 失访 | 失访 |
| 21 | 女 | 62 | 皮肤、头顶部 | 发现肿块 | 手术 | 失访 | 失访 |
| 22 | 男 | 14 | 眼眶 | 发现肿块 | 手术 | 失访 | 失访 |
| 23 | 女 | 37 | 颏下淋巴结 | 发现肿块 | 手术 | 失访 | 失访 |
注:CHOP方案:环磷酰胺750 mg/m2,第1天;表柔比星80 mg/m2,第1天;长春地辛4 mg,第1天;泼尼松100 mg/d,第1~5天
CT或MRI示本组23例RDD表现为淋巴结肿大,或淋巴结外实性占位,可伴有强化(图1A, 图1B, 图1C)。
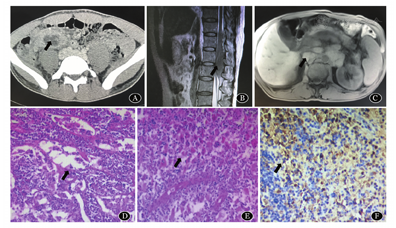
A:CT示结肠肿块较明显强化,周围可见多枚肿大淋巴结;B:MRI示胸12-腰2水平椎管内肿块明显强化;C:MRI T2像可见肿大淋巴结呈串珠样改变;D:淋巴结病理形态示淋巴结淋巴窦扩张,窦内可见大量组织细胞增生(HE染色,× 400);E:淋巴结外病理形态示组织细胞吞噬淋巴细胞现象(HE染色,× 400);F:免疫组化示组织细胞S100蛋白阳性(× 400)
RDD结外病变镜下主要表现为软组织中出现明暗相间的结构,明区主要为增生的组织细胞,细胞体积大,核空泡状,可见明显吞噬淋巴细胞,浆细胞现象,主要表现为"吞入",其内淋巴细胞、浆细胞轮廓清晰;暗区主要为大量淋巴细胞,浆细胞浸润(图1D)。皮肤型结外RDD有特殊的病理特点,病变组织主要集中在皮肤真皮或皮下软组织中,可见大量成片的淡染组织细胞增生伴吞噬淋巴细胞现象,缺乏窦样分布的特点,背景中见团块状深染的淋巴细胞及浆细胞浸润。RDD淋巴结病变镜下可见淋巴结结构存在,淋巴窦显著扩张,其内大量组织细胞增生,可见明显吞噬淋巴细胞、浆细胞现象(图1E),RDD后期表现为组织细胞减少,纤维组织增生,尤其以结外病变表现更明显,但仍可见多少不等的浆细胞和淋巴细胞,可提供一定的诊断线索。免疫组化显示组织细胞87%呈S100阳性(20/23,图1F),87%呈CD68阳性(20/23),100%呈CD163阳性(23/23),100%呈CD1a阴性(23/23)。
本组9例RDD患者WES原始数据采用Trimmomatic、bwa、GATK软件进行数据比对及变异分析,包括单核苷酸变异(SNV)或碱基序列的插入与缺失(InDel)等。采用Annovar软件对变异位点进行注释,并对变异位点进行检索数据库及相关文献。注释数据库如下:人类基因参考组(Hg38)、dbSNP、cytoBand、genomicSuperDups、avsnp150、ljb26_all、1000g2015aug_all、COSMIC83、ExAC03和ClinVar_20170905,同时应用生物信息学软件PolyPhen.2、SIFT对获得的位点进行致病性预测。一共发现16种基因突变,包括TTN、KMT2D、NOTCH1、FAT1、MFHAS1、NCOR1、MEF2B、mTOR、PLCG2、NFKBIE、EPPK1、TET2、RHOA、ATG2B、LYN、NRXN3等。合计25个SNV或InDel(22个错义突变,1个无义突变,1个移码缺失,1个非移码插入),详细结果见表2。
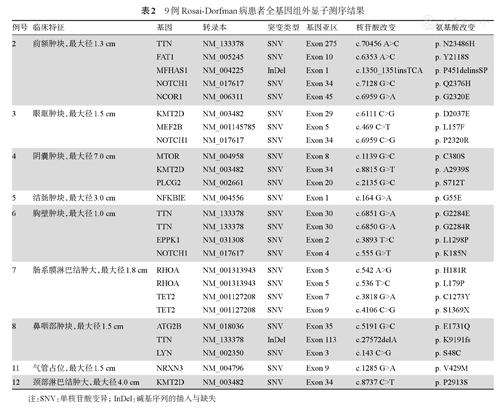
9例Rosai-Dorfman病患者全基因组外显子测序结果
9例Rosai-Dorfman病患者全基因组外显子测序结果
| 例号 | 临床特征 | 基因 | 转录本 | 突变类型 | 基因亚区 | 核苷酸改变 | 氨基酸改变 |
|---|---|---|---|---|---|---|---|
| 2 | 前额肿块,最大径1.3 cm | TTN | NM_133378 | SNV | Exon 275 | c.70456 A>C | p. N23486H |
| FAT1 | NM_005245 | SNV | Exon 10 | c.6353 A>C | p. Y2118S | ||
| MFHAS1 | NM_004225 | InDel | Exon 1 | c.1350_1351insTCA | p. P451delinsSP | ||
| NOTCH1 | NM_017617 | SNV | Exon 34 | c.7128 G>C | p. Q2376H | ||
| NCOR1 | NM_006311 | SNV | Exon 45 | c.6959 G>A | p. G2320E | ||
| 3 | 眼眶肿块,最大径1.5 cm | KMT2D | NM_003482 | SNV | Exon 29 | c.6111 C>G | p. D2037E |
| MEF2B | NM_001145785 | SNV | Exon 5 | c.469 C>T | p. L157F | ||
| NOTCH1 | NM_017617 | SNV | Exon 34 | c.6959 C>G | p. P2320R | ||
| 4 | 阴囊肿块,最大径7.0 cm | MTOR | NM_004958 | SNV | Exon 8 | c.1139 G>C | p. C380S |
| KMT2D | NM_003482 | SNV | Exon 34 | c.8815 G>T | p. A2939S | ||
| PLCG2 | NM_002661 | SNV | Exon 20 | c.2135 G>C | p. S712T | ||
| 5 | 结肠肿块,最大径3.0 cm | NFKBIE | NM_004556 | SNV | Exon 1 | c.164 G>A | p. G55E |
| 6 | 胸壁肿块,最大径1.0 cm | TTN | NM_133378 | SNV | Exon 30 | c.6851 G>A | p. G2284E |
| TTN | NM_133378 | SNV | Exon 30 | c.6850 G>A | p. G2284R | ||
| EPPK1 | NM_031308 | SNV | Exon 2 | c.3893 T>C | p. L1298P | ||
| NOTCH1 | NM_017617 | SNV | Exon 4 | c.555 G>T | p. K185N | ||
| 7 | 肠系膜淋巴结肿大,最大径1.8 cm | RHOA | NM_001313943 | SNV | Exon 5 | c.542 A>G | p. H181R |
| RHOA | NM_001313943 | SNV | Exon 5 | c.536 T>C | p. L179P | ||
| TET2 | NM_001127208 | SNV | Exon 7 | c.3818 G>A | p. C1273Y | ||
| TET2 | NM_001127208 | SNV | Exon 9 | c.4106 C>G | p. S1369X | ||
| 8 | 鼻咽部肿块,最大径1.5 cm | ATG2B | NM_018036 | SNV | Exon 35 | c.5191 G>C | p. E1731Q |
| TTN | NM_133378 | InDel | Exon 113 | c.27572delA | p. K9191fs | ||
| LYN | NM_002350 | SNV | Exon 3 | c.143 C>G | p. S48C | ||
| 11 | 气管占位,最大径1.5 cm | NRXN3 | NM_004796 | SNV | Exon 9 | c.1285 G>A | p. V429M |
| 12 | 颈部淋巴结肿大,最大径4.0 cm | KMT2D | NM_003482 | SNV | Exon 34 | c.8737 C>T | p. P2913S |
注:SNV:单核苷酸变异;InDel:碱基序列的插入与缺失
本组所有患者均经手术切除病灶,1例(例7)患者术后予2个疗程CHOP方案(环磷酰胺750 mg/m2,第1天;表柔比星80 mg/m2,第1天;长春地辛4 mg,第1天;泼尼松100 mg/d,第1~5天)化疗巩固。4例失访患者无法评价疗效,余19例患者中位随访24(1~67)个月,疾病均无复发。
RDD是一种较为少见的组织细胞增生性疾病,发病率约为0.005%,平均发病年龄约20.6岁,各个年龄段均可发病[1]。RDD根据病变累及范围可分为3个类型,淋巴结型、结外型和混合型。在组织细胞学会工作组(Working Group of the Histiocyte Society,HS)2016年版组织细胞和肿瘤修订分类中属于R组,又可进一步分为家族型、经典型(淋巴结型)、结外型、肿瘤相关型和自身免疫疾病相关型[2]。
Rosai和Dorfman于1990年首次报道了423例RDD,其中43%出现淋巴结外受累,单一结外受累仅占5%[3]。1996年Hammond等[4]报道淋巴结外受累占40%,23%为结外单一病灶。近年来,随着疾病诊断水平的提高,结外受累及单一结外病变的比例逐渐增加。MD Anderson癌症中心报道指出RDD淋巴结外受累占76%,62%为结外单一病灶,国内相似的报道分别为92%和77%[5,6]。本组23例RDD患者,87%(20/23)为结外受累,单一结外受累(19/23)达83%,与近年RDD构成比例基本相符。值得注意的是,本组病例中有9例单独发生在皮肤,占结外病变的45%(9/20),高于国外文献报道的10%[7]。
RDD的病因尚不明确,可能与病毒感染或自身免疫异常相关,但均未得到证实。本组病例中均无家族史和自身免疫疾病史。既往认为RDD不存在BRAF-V600E突变,且BRAF-V600E突变有助于鉴别RDD和ECD或LCH。最近Fatobene等[8]在1例RDD患者中发现BRAF-V600E突变,而本组病例中均未检出BRAF-V600E突变。RDD中是否存在BRAF-V600E突变及其鉴别诊断意义还有待进一步扩大样本进行观察。此外,文献报道RDD中还发现TNFRSF6、NRAS、KRAS、MAP2K1、PIK3CA和ARAF基因突变,以及在家族型RDD中存在SLC29A3突变[5,9,10,11,12,13]。
9例患者进行全基因组外显子测序。结果共发现16个基因突变,25个位点发生SNV或InDel,在RDD患者中均为首次报道,临床意义尚不明确。①1例患者发现TET2基因2个位点发生突变,分别为7号外显子上错义突变,c.3818G>A(p.C1273Y),9号外显子上无义突变,stopgain c.4106C>G(p.S1369X)。TET2基因被认为是一种抑癌基因,通过影响DNA去甲基化和组蛋白糖基化的双表观遗传修饰调控基因转录,参与调控造血组织。TET2基因突变见于92%的血管免疫母细胞性淋巴瘤(AITL)[14],14%的成人T细胞白血病/淋巴瘤(ATLL)[15]。② 1例RDD患者mTOR基因8号外显子上发现错义突变,c.1139G>C(p. C380S)。PI3K/雷帕霉素靶蛋白(mTOR)信号通路在体内广泛存在,调控细胞的生长与代谢,并且在多种恶性肿瘤中存在高表达和过度活化。Cooper等[16]报道了1例RDD患者使用雷帕霉素治疗且达到了完全缓解,提示PI3K/mTOR信号通路在RDD中可能存在异常。本组该患者同时存在KMT2D、PLCG2基因突变。③3例患者(33%)检测到KMT2D基因错义突变,分别为:29号外显子c.6111C>G(p.D2037E);34号外显子c.8815G>T(p.A2939S)和9号外显子c.1285G>A(p. V429M)。KMT2D编码赖氨酸特异性甲基转移酶,参与组蛋白甲基化修饰的过程,是一种肿瘤抑制因子,在调节增强子功能中起关键作用。在动物实验中证实KMT2D基因失活加速B细胞淋巴瘤小鼠模型肿瘤的发生[17]。④3例患者(33%)存在NOTCH1基因错义突变,分别为:34号外显子c.7128G>C(p.Q2376H)和c.6959C>G (p.P2320R);4号外显子c.555G>T(p.K185N)。NOTCH1信号通路广泛参与细胞的分化、增殖和凋亡等过程,在多种血液系统恶性肿瘤和其他实体肿瘤中异常激活,并且与PI3K/mTOR信号通路存在密切的相互作用。Xerri等[18]对22例LCH及3例郎格汉斯组织细胞肉瘤(Langerhans cell sarcoma,LCS)病变组织进行二代测序,3例LCS中均测出KMT2D、NRAS、MAP2K1基因突变,2例LCS中测出NOTCH1基因突变,而在LCH中没有发现KMT2D、NRAS和NOTCH1突变,多基因突变可能是郎格汉斯细胞肿瘤侵袭行为的基础。简言之,9例RDD患者均检测出基因突变,其中5例同时存在3种及以上的基因突变,提示多基因突变可能通过复杂的相互作用共同导致RDD发生。这些突变的具体临床意义和预后之间的关系还需要进一步研究。此外我们并没有发现既往文献报道的NRAS、KRAS、PIK3CA、TNFRSF6、MAP2K1、BRAF、ARAF及SLC29AC等突变[5,8,9,10,11,13]。
一般认为RDD属于自限性疾病,多数可在数月至数年内自行消退,仅有少数病例复发或导致死亡。治疗上,无论是结内或结外RDD,对单发病例多主张手术切除,国外文献报道持续缓解率为80%~100%[7,19,20,21,22]。免疫调节剂沙利度胺和来那度胺对复发难治RDD有效[23]。Pulsoni等[24]报道放疗治愈率为33.3%。Jacobsen等[25]报道1例对糖皮质激素治疗无反应的RDD,使用二代测序发现KRAS基因突变,患者接受MEK抑制剂cobimetinib治疗2个月达到缓解。Lee等[26]观察到经过治疗的RDD患者血清IL-6水平较前可有明显下降,对1例初始予糖皮质激素联合长春新碱治疗无反应的患者使用IL-6拮抗剂siltuximab,治疗1个周期后患者达到完全缓解。本组23例RDD均行手术切除,仅1例患者术后予CHOP方案2个疗程巩固,19例患者中位随访24个月,患者一般情况可,均未见复发。
总之,RDD属于良性组织细胞增生性疾病,临床过程属于自限性,形态学表现具有特征性,部分病例可能存在克隆性基因改变。本研究我们发现部分RDD存在mTOR、KMT2D和NOTCH1基因改变,有一定深入探讨价值。

























