
以内质网应激(ERS)为切入点,深入研究全反式维甲酸(ATRA)调控FLT3-ITD突变蛋白表达下降,导致FLT3-ITD突变阳性白血病细胞凋亡的分子机制。
ATRA处理FLT3-ITD突变阳性白血病细胞株(MV4-11和MOLM13),流式细胞术检测细胞凋亡,实时荧光定量PCR和Western blot法分别检测细胞ERS相关和(或)自噬相关基因及蛋白的表达。
低剂量ATRA可提高FLT3-ITD突变阳性白血病细胞的ERS水平;ATRA作用于ERS相关的PERK/eif2ɑ信号通路,使FLT3-ITD突变阳性细胞的ERS水平持续升高,CHOP基因表达上调;FLT3-ITD突变阳性细胞经ATRA处理后,FLT3-ITD蛋白表达水平降低;细胞内与ERS有关的并且主要的两个蛋白降解途径中,与内质网关联降解(ERAD)相关的蛋白ATF6表达无明显变化,而与内质网激活自噬(ERAA)相关的自噬相关蛋白Atg5和Atg7表达明显升高。
ATRA通过激活PERK/eif2ɑ信号通路持续上调FLT3-ITD突变阳性细胞的ERS水平,通过ERAA途径自噬降解FLT3-ITD蛋白,促进白血病细胞凋亡。研究结果为临床应用ATRA治疗难治性FLT3-ITD突变阳性白血病提供了初步实验依据。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
FMS样酪氨酸激酶3(FMS-like tyrosine 3,FLT3)属于Ⅲ型酪氨酸受体,在急性髓系白血病(AML)患者中高表达且部分发生基因突变[1]。其中FLT3内部串联重复(FLT3-ITD)突变最常见[2],具有更高的致癌潜能[3],导致疗效差和易复发[4],是难治性AML的独立高危因素[5,6]。靶向降低FLT3-ITD表达是治疗该类难治性白血病的关键环节。
内质网应激(endoplasmic reticulum stress,ERS)是指细胞受应激事件影响时,蛋白的翻译、转运、加工和分泌出现异常,错误折叠蛋白在内质网蓄积,无法向高尔基体和细胞膜转运,导致内质网压力升高,其形态和功能发生改变。ERS激活的错误折叠蛋白降解主要有内质网关联的降解(ER-associated degradation,ERAD)和内质网激活的自噬(ER-activated autophagy,ERAA)途径[7,8,9],前者与ATF6和IRE1ɑ通路介导的蛋白酶体途径相关,后者与PERK和(或)IRE1ɑ通路或钙离子介导的巨自噬途径相关[7,10]。当ERS持续且无法缓解时,细胞发生凋亡[11]。
研究表明,肿瘤细胞的ERS水平较正常细胞高,有利于维持其生存[12,13,14]。FLT3蛋白发生ITD突变后,自发形成同源二聚体,蓄积在内质网腔内[15],FLT3-ITD突变阳性AML细胞的ERS水平可能较高。本研究组前期研究显示,全反式维甲酸(ATRA)可诱导FLT3-ITD蛋白表达降低并促进白血病细胞凋亡[16],但机制未明。有报道显示,ATRA能提高急性早幼粒细胞白血病(APL)细胞的ERS水平[17]。据此推测ATRA增强FLT3-ITD突变阳性AML细胞的ERS,是导致FLT3-ITD蛋白表达降低和诱导FLT3-ITD突变阳性白血病细胞凋亡的重要机制之一。本研究以FLT3-ITD突变阳性AML细胞株MV4-11和MOLM13为模型,探究ATRA诱导FLT3-ITD蛋白表达降低、促进FLT3-ITD突变白血病细胞凋亡是否与ERS相关,为FLT3-ITD突变AML的治疗提供新思路。
实验用的细胞株均为人类白血病细胞株,MV4-11和MOLM13为FLT3-ITD突变阳性AML细胞株,RS4-11、THP1和HL-60为FLT3-ITD突变阴性细胞株。其中MOLM13购自德国DSMZ细胞库,其他细胞株购自美国模式培养物研究所(ATCC)或中国医学科学院血液学研究所。MV4-11细胞株用含10%胎牛血清的IMDM培养液,其他细胞株用含10%胎牛血清的RPMI 1640培养液,于含5%的CO2、37 ℃培养箱中培养,根据细胞增殖情况定期更换培养液。
ATRA(美国Sigma-Aldrich公司产品,货号R2625)溶于DMSO保存。4-PBA、GSK2606414和Guanabenz购自索拉宝公司,溶于DMSO保存,DMSO在培养基的终浓度<0.1%。PERK、p-PERK、BiP、eif2ɑ、p-eif2ɑ、Atg5和Atg7抗体购自美国Cell Signaling Technology公司。
用Annexin Ⅴ-FITC/PI凋亡检测试剂盒[东仁化学科技(上海)有限公司产品]检测细胞凋亡。取对数生长期的细胞进行细胞计数,用培养基调整细胞密度为1×105/L,接种于12孔板内,予不同浓度的ATRA处理。处理后避光培养24、48及72 h,分别收集不同培养时间的细胞离心(1000 r/min,10 min)。弃上清,4 ℃预冷的PBS洗涤细胞2次后用结合缓冲液制成细胞悬液。依次加入Annexin Ⅴ-FITC和PI各5 μl,吹打混匀,室温下避光孵育15 min。在2 h内用流式细胞仪完成各组细胞凋亡率的检测。
用TRIzol法提取总RNA。将细胞离心后弃上清,裂解于TRIzol试剂,氯仿将RNA分离后用75%乙醇和无水乙醇各洗涤2次。真空抽干乙醇溶液,将RNA溶解于DEPC水,检测RNA的浓度和纯度。去除基因组DNA以及cDNA的合成采用Promage RR047a试剂盒。RT-qPCR以cDNA为模板扩增目的基因和内参基因。引物:GAPDH上游引物:5′-GCCAACACAGTGCTGTCTGG-3′;下游引物:5′-GCTCAGGAGGAGCAATGATCTTG-3′。BiP上游引物:5′-CTCCTGAAGGGGAACGTCTG-3′;下游引物:5′-CCACCTTGAACGGCAAGAAC-3′。CHOP上游引物:5′-TCCAGCCACTCCCCATTATC-3′;下游引物:5′-GCAGGGTCAAGAGTGGTGAA-3′。XBP1-s上游引物:5′-CTGAGTCCGCAGCAG GTG-3′;下游引物:5′-AGGGAGGCTGGTAA GGAACT-3′。XBP1-u上游引物:5′-CAGACTACG TGCACCTCTGC-3′;下游引物:5′-GGGTCCTTCT GGGTAGACCT-3′。
ATRA避光处理细胞24、48和72 h后,分别收集、洗涤和裂解细胞,提取细胞总蛋白及胞质蛋白进行定量和热变性处理。根据目的蛋白大小配制不同浓度的十二烷基硫酸钠-聚丙烯酰胺凝胶进行电泳(SDS-PAGE),根据蛋白大小选择合适的转膜条件将蛋白转移到硝酸纤维素(NC)膜上,加入一抗在4 ℃过夜孵育印迹,加入辣根过氧化物酶(HRP)耦联的二抗孵育1 h,用增强型化学发光法(ECL)检测确定目的蛋白的表达强弱,GAPDH或β-actin作为内参,确保每个泳道的总蛋白及胞质蛋白上样量一致。
实验数据均来自于3次或3次以上结果相似的独立实验,计量资料数据采用均数±标准差表示,采用SPSS 22.0软件进行数据分析,采用直方图和Kolmogorov-Smirnov检验正态性,对每组数据进行方差齐性检验,两独立样本均数比较采用Student’st检验,多个样本均数比较采用方差分析,P<0.05为差异有统计学意义,统计图采用Graphpad Prism 8.0进行绘制。
不同浓度的ATRA(0、0.5和1.0 μmol/L)作用于MV4-11、MOLM13和RS4-11细胞48和72 h,流式细胞术检测细胞凋亡情况。结果显示,ATRA对MV4-11和MOLM13细胞的促凋亡作用呈现明显的时间依赖性和剂量依赖性,其中低剂量ATRA(0.5 μmol/L)在72 h已有明显的促凋亡作用(图1A、图1B)。而ATRA对RS4-11细胞的促凋亡作用较弱(图1C)。提示ATRA选择性地杀伤FLT3-ITD突变阳性AML细胞。
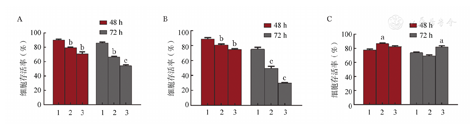
A、B分别为FLT3-ITD突变阳性细胞MV4-11和MOLM13;C为FLT3-ITD突变阴性细胞RS4-11。1:空白对照组;2:0.5 μmol/L ATRA作用组;3:1.0 μmol/L ATRA作用组。与空白对照组比较,aP<0.05,bP<0.01,cP<0.001
BiP和XBP1-s上调是ERS的生物学标志。将不同浓度的ATRA(0、0.5和1.0 μmol/L)作用于MV4-11、MOLM13、RS4-11和HL-60细胞24、48及72 h,RT-qPCR检测ERS标志基因BiP和XBP1-s的表达。结果显示,与RS4-11和HL-60细胞比较,MV4-11和MOLM13细胞在ATRA作用下BiP基因的表达明显升高,MOLM13细胞XBP1-s基因的表达亦明显升高。与空白对照组相比,差异具有统计学意义(P值均<0.05),其中0.5 μmol/L ATRA对ERS水平上调作用最明显(表1)。提示低剂量ATRA可显著上调FLT3-ITD突变阳性AML细胞的ERS水平。
全反式维甲酸(ATRA)对FLT3-ITD突变阳性和阴性白血病细胞株细胞内质网应激水平的影响( ±s,n=3)
±s,n=3)
全反式维甲酸(ATRA)对FLT3-ITD突变阳性和阴性白血病细胞株细胞内质网应激水平的影响(±s,n=3)
| 组别 | MV4-11细胞 | MOLM13细胞 | |||||
|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | ||
| BiP基因 | |||||||
| 空白对照组 | 1.00±0.15 | 1.00±0.08 | 1.00±0.08 | 1.00±0.09 | 1.00±0.06 | 1.00±0.05 | |
| 0.5 μmol/L ATRA组 | 1.74±0.15b | 2.33±0.13c | 2.50±0.11c | 1.49±0.05b | 2.24±0.09c | 2.41±0.12c | |
| 1.0 μmol/L ATRA组 | 1.59±0.20a | 1.83±0.30b | 2.00±0.04c | 1.59±0.05c | 2.32±0.11c | 3.19±0.05c | |
| XBP1-s基因 | |||||||
| 空白对照组 | 1.00±0.08 | 1.00±0.12 | 1.00±0.04 | 0.98±0.06 | 1.07±0.11 | 1.10±0.11 | |
| 0.5 μmol/L ATRA组 | 0.81±0.07 | 1.12±0.14 | 0.87±0.04 | 1.22±0.08a | 2.22±0.26b | 1.74±0.14b | |
| 1.0 μmol/L ATRA组 | 0.72±0.02 | 1.01±0.15 | 0.61±0.02 | 1.41±0.07b | 2.36±0.28b | 2.34±0.21c | |
| 组别 | RS4-11细胞 | HL-60细胞 | |||||
|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | ||
| BiP基因 | |||||||
| 空白对照组 | 1.00±0.36 | 1.00±0.14 | 1.00±0.07 | 1.00±0.03 | 1.00±0.15 | 1.00±0.14 | |
| 0.5 μmol/L ATRA组 | 1.14±0.11 | 1.06±0.09 | 1.09±0.07 | 1.12±0.06 | 0.91±0.08 | 1.49±0.05 | |
| 1.0 μmol/L ATRA组 | 1.17±0.07 | 1.19±0.06 | 1.12±0.04 | 1.21±0.08 | 1.28±0.05 | 1.57±0.12 | |
| XBP1-s基因 | |||||||
| 空白对照组 | 1.00±0.10 | 1.00±0.11 | 1.00±0.13 | 1.00±0.15 | 1.00±0.17 | 1.00±0.17 | |
| 0.5 μmol/L ATRA组 | 1.48±0.38 | 1.12±0.14 | 1.41±0.24 | 0.76±0.24 | 0.43±0.08b | 0.63±0.11a | |
| 1.0 μmol/L ATRA组 | 1.34±0.25 | 1.25±0.12 | 1.18±0.17 | 0.69±0.14 | 0.36±0.06b | 0.67±0.07a | |
注:表中的数据为BiP和XBP1-s基因的相对表达量,内参基因为GAPDH。与空白对照组比较,aP<0.05,bP<0.005,cP<0.001
为进一步明确低剂量ATRA调控FLT3-ITD突变阳性细胞ERS的信号通路,我们检测了参与持续ERS最重要的PERK/eif2ɑ通路。Western blot检测结果显示,0.5 μmol/L ATRA作用于细胞72 h后,MV4-11和MOLM13细胞的PERK磷酸化蛋白表达增加,RS4-11和THP1细胞的PERK磷酸化蛋白表达无明显变化(图2A);另外,ATRA处理后MV4-11和MOLM13细胞的BiP蛋白表达上升,eif2ɑ蛋白磷酸化水平降低(图2B)。
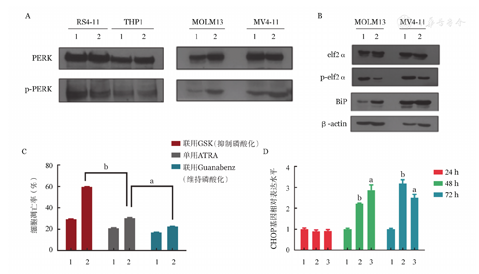
A:ATRA作用于FLT3-ITD突变阳性和阴性白血病细胞株后PERK及其磷酸化蛋白的表达(1:空白对照组;2:ATRA作用组);B:ATRA作用于FLT3-ITD突变阳性细胞株后BiP和eif2ɑ及其磷酸化蛋白的表达变化(1:空白对照组;2:ATRA作用组);C:干预eif2ɑ蛋白的磷酸化后ATRA处理MV4-11细胞72 h对细胞凋亡率的影响(1:空白对照组;2:ATRA作用组。与单用ATRA组比较,aP<0.05,bP<0.001);D:ATRA作用于MV4-11细胞后CHOP基因的表达情况(1:空白对照组;2:0.5 μmol/L ATRA作用组;3:1.0 μmol/L ATRA作用组。与空白对照组比较,aP<0.005,bP<0.001)
进一步使用GSK2606414(简称GSK,抑制eif2ɑ磷酸化)和Guanabenz(维持eif2ɑ磷酸化)分别干预eif2ɑ的磷酸化。结果发现,将ATRA(0.5 μmol/L)与GSK(1.0 μmol/L)共处理MV4-11细胞72 h,其细胞凋亡率较单用ATRA时升高;而ATRA(0.5 μmol/L)与Guanabenz(17 μmol/L)共处理MV4-11细胞72 h,其细胞凋亡率较单用ATRA时下降。采用单因素方差分析不同处理组MV4-11细胞凋亡率,其差异具有统计学意义(F=1656,P<0.001)。以单用ATRA组为对照,采用Dunnett's法两两比较,结果差异均具有统计学意义(P值均<0.05)(图2C)。表明维持eif2ɑ磷酸化可减弱ATRA的作用,ATRA通过抑制eif2ɑ磷酸化作用于FLT3-ITD突变白血病细胞。RT-qPCR检测PERK通路下游与凋亡相关的靶基因CHOP,结果发现ATRA处理后的MV4-11细胞CHOP基因表达明显上调(图2D)(P<0.05)。
4-苯基丁酸钠盐(4-PBA)是常用的ERS抑制剂。为探究ERS、ATRA和FLT3-ITD蛋白表达三者的关系,将MV4-11细胞分为空白对照组、ATRA组和ATRA+4-PBA组,ARTA和4-PBA的剂量分别为0.5 μmol/L和2.5 mmol/L,细胞处理后避光培养72 h。Western blot检测结果显示:①与空白对照组比较,ATRA组的FLT3-ITD蛋白表达水平明显降低,而ATRA+ 4-PBA组的FLT3-ITD蛋白表达水平高于ATRA组,但较空白对照组低(图3A);②ATRA处理后,各组ERAD途径相关的ATF6蛋白表达量无明显变化(图3B)。另外,进一步检测ATRA处理后MV4-11和MOLM13细胞的ERAA途径相关的自噬标志物Atg5和Atg7的蛋白表达情况,Western blot结果显示,与空白对照组相比,ATRA组的Atg5和Atg7蛋白表达增高(图3C)。提示ATRA通过ERAA途径诱导FLT3-ITD蛋白自噬降解。
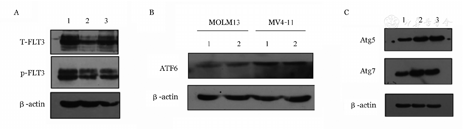
A:ERS水平对ATRA降低MV4-11细胞FLT3-ITD蛋白表达作用的影响(1:空白对照组;2:ATRA作用组;3:ATRA+4-PBA作用组);B:ATRA对FLT3-ITD突变阳性AML细胞ATF6蛋白表达的影响(1:空白对照组;2:ATRA作用组);C:ATRA对FLT3-ITD突变阳性MV4-11细胞自噬相关蛋白Atg5和Atg7表达的影响(1:空白对照组;2:ATRA作用48 h组;3:ATRA作用72 h组)
本研究在前期研究[16]的基础上,从ERS的角度深入探讨ATRA通过降低FLT3-ITD蛋白表达促进FLT3-ITD突变阳性白血病细胞凋亡的机制。结果显示,ATRA通过激活PERK/eif2ɑ信号通路持续上调FLT3-ITD突变阳性细胞的ERS水平,通过ERAA途径诱导FLT3-ITD突变蛋白自噬降解,最终促进白血病细胞凋亡。
与正常细胞比较,多数肿瘤细胞ERS水平升高[12]。研究表明,蛋白发生突变后,构象不稳定,在内质网中容易形成低聚物或聚合物,蓄积在内质网造成ERS[7]。Chunaram等[15]研究FLT3突变蛋白在细胞的分布发现,FLT3发生ITD突变后,可自发形成同源二聚体,并蓄积在内质网腔内,FLT3-ITD突变阳性AML细胞的ERS水平可能较高。
临床上可利用肿瘤细胞ERS水平较高这一特点,用于肿瘤的治疗。研究显示,维莫非尼(vemurafenib)可通过提高ERS水平诱导BRAFV600E基因突变的黑色素瘤细胞凋亡[18]。ATRA常用于APL的治疗[19],其主要机制是靶向融合基因PML-RARɑ中的RARɑ部分,降解PML-RARɑ蛋白,恢复野生型PML和RARɑ基因的功能,诱导APL细胞分化成熟[20]。近年研究还发现,ATRA能提高APL细胞的ERS[17]。
ERS过程中主要激活非折叠蛋白反应(unfolding protein respone,UPR),包括PRRK、ATF6和IRE1ɑ通路,通过抑制翻译、协助蛋白正确折叠和激活蛋白发生泛素化和自噬降解,缓解ERS。细胞在非ERS时,PERK、ATF6和IRE1ɑ与免疫球蛋白结合分子(BIP/Grp78)结合,维持信号通路的非活化;ERS时UPR信号通路被激活,BIP与PERK、ATF6和IRE1ɑ分离且表达上调,XBP1-s表达增加,PERK和ATF6通路以及XBP1-s可进一步促进BIP的表达[21,22]。因此BIP和XBP1-s常作为评估ERS水平的重要因子。本研究通过实验检测ATRA处理后白血病细胞BIP和XBP1-s的mRNA相对表达量变化情况,结果发现ATRA可上调FLT3-ITD突变阳性的白血病细胞ERS,但对FLT3-ITD突变阴性的白血病细胞ERS无明显上调作用。证明ATRA可选择性地上调FLT3-ITD突变阳性的AML细胞系的ERS水平。
为此,我们提出一个问题:ATRA调控FLT3-ITD白血病细胞ERS的信号通路是什么?有研究发现当细胞处于持续ERS时,PERK/eif2α通路起关键作用[23]。当PERK磷酸化激活后,eif2ɑ发生磷酸化改变,主要作用有:①抑制细胞大部分蛋白的翻译速率,缓和ERS;②随后恢复ATF4蛋白表达,激活GADD34表达,eif2ɑ进入去磷酸化状态,恢复胞内蛋白合成[24,25],该负反馈机制在ERS缓解时起恢复细胞正常活动的作用。当内质网压力持续且无法缓解时,PERK/eif2ɑ持续激活[23],下游凋亡相关的靶基因CHOP大量表达,细胞稳态失衡,发生凋亡[11]。本研究显示,ATRA选择性地增强FLT3-ITD突变阳性AML细胞的PERK磷酸化,BiP表达上调,提高ERS水平。另一方面,ATRA抑制eif2ɑ磷酸化,使得细胞内蛋白继续大量合成,ERS持续存在且难以缓解。我们在研究中还发现,ATRA使FLT3-ITD AML细胞的PERK通路下游促凋亡基因CHOP基因表达明显上调,提示其诱导的细胞凋亡与ERS水平持续升高有关。
本课题组前期研究显示,ATRA可降低FLT3-ITD突变蛋白表达[16],但机制有待进一步阐明。有研究报道,ERS可激活细胞内蛋白包括错误蛋白的降解,主要有ERAD和ERAA途径,前者与ATF6和IRE1ɑ通路介导的蛋白酶体途径相关,后者与PERK和(或)IRE1ɑ通路或钙离子介导的巨自噬途径相关[7]。本研究发现,ATRA可降低FLT3-ITD蛋白的表达,其作用在高水平的ERS细胞内环境下更为明显,但主要通过ERAA通路相关的巨自噬而不是ERAD相关的泛素化-蛋白酶体途径降解。有研究表明[7],蛋白发生突变后,构象不稳定,在内质网中容易形成低聚物或聚合物,蓄积在内质网内难以发生泛素化,较难通过ERAD途径(蛋白酶体相关)降解。FLT3-ITD突变蛋白与野生型FLT3蛋白不同,会自发形成同源二聚体[15],可解释FLT3-ITD主要不是通过ERAD途径降解的现象。ERAA是与PERK通路相关的巨自噬途径[7],本研究显示,ATRA促进PERK磷酸化,激活PERK通路,并使自噬标志蛋白Atg5和Atg7[26]表达上升,而与ERAD相关的ATF6蛋白无明显变化,提示ATRA降低FLT3-ITD蛋白的表达,与其上调的ERS水平相关,且主要通过激活ERAA途径自噬降解FLT3-ITD。
综上,ATRA可选择性地上调FLT3-ITD突变阳性AML细胞的ERS水平,导致FLT3-ITD自噬降解,最终促进白血病细胞凋亡。在ERS状态下,ATRA的重要作用靶点是ERS的关键通路——PERK/eif2ɑ通路,通过促进PERK磷酸化激活,抑制eif2ɑ磷酸化,使FLT3-ITD突变阳性AML细胞的ERS持续处于高水平,激活ERAA途径自噬降解FLT3-ITD蛋白,最终导致白血病细胞凋亡。本研究初步揭示了ATRA通过调控ERS诱导FLT3-ITD蛋白自噬降解促进白血病细胞凋亡的分子机制,为今后将ATRA用于临床治疗难治性FLT3-ITD突变阳性AML和改善预后提供初步实验依据。

























